ジャーナル: Cell Rep / 年: 2022 タイトル: A neutralizing epitope on the SD1 domain of SARS-CoV-2 spike targeted following infection and vaccination. 著者: Jeffrey Seow / Hataf Khan / Annachiara Rosa / Valeria Calvaresi / Carl Graham / Suzanne Pickering / Valerie E Pye / Nora B Cronin / Isabella Huettner / Michael H Malim / Argyris Politis / ...著者: Jeffrey Seow / Hataf Khan / Annachiara Rosa / Valeria Calvaresi / Carl Graham / Suzanne Pickering / Valerie E Pye / Nora B Cronin / Isabella Huettner / Michael H Malim / Argyris Politis / Peter Cherepanov / Katie J Doores / 要旨: Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) spike is the target for neutralizing antibodies elicited following both infection and vaccination. While extensive research has shown that ...Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) spike is the target for neutralizing antibodies elicited following both infection and vaccination. While extensive research has shown that the receptor binding domain (RBD) and, to a lesser extent, the N-terminal domain (NTD) are the predominant targets for neutralizing antibodies, identification of neutralizing epitopes beyond these regions is important for informing vaccine development and understanding antibody-mediated immune escape. Here, we identify a class of broadly neutralizing antibodies that bind an epitope on the spike subdomain 1 (SD1) and that have arisen from infection or vaccination. Using cryo-electron microscopy (cryo-EM) and hydrogen-deuterium exchange coupled to mass spectrometry (HDX-MS), we show that SD1-specific antibody P008_60 binds an epitope that is not accessible within the canonical prefusion states of the SARS-CoV-2 spike, suggesting a transient conformation of the viral glycoprotein that is vulnerable to neutralization.
The micrograph stacks were aligned, binned to the physical pixel size of 1.1 A and summed, with dose weighting applied, as implemented in MotionCor2
粒子像選択
選択した数: 1740430 詳細: Initially, particles were picked with Gautomatch-v0.53 using 2D class averages of the trimeric spike, low-pass filtered to 20 A resolution, as templates. The resulting 1,740,430 particles, ...詳細: Initially, particles were picked with Gautomatch-v0.53 using 2D class averages of the trimeric spike, low-pass filtered to 20 A resolution, as templates. The resulting 1,740,430 particles, extracted in Relion-3.1 and binned to a pixel size of 4.4 A, were subjected to two rounds of reference-free 2D classification in cryoSPARC-2. 283,956 particles belonging to well-defined 2D classes were subjected to classification into twelve 3D classes in Relion-3.1. Neither the 2D nor 3D class averages of the trimeric spike revealed features attributable to a bound Fab molecule. Next, 3,772,722 particles were picked using 2D class averages of the dissociated spikes. Following two rounds of 2D classification in cryoSPARC-2, 753,837 particles, re-extracted with pixel size of 2.2 A, were subjected to 3D classification in Relion-3.1 into 9 classes using an initial model obtained by Ab-initio reconstruction in cryoSPARC-2. The procedure revealed a single well-defined 3D class containing 208,343 particles (27.4%) of S1 protein with a bound Fab molecule. The particles, re-extracted without binning (with a pixel size 1.1 A), were subjected to two rounds of 3D classification using Ab-initio reconstruction in cryoSPARC-2 with 2 classes and class similarity set to 0. At the end of each round, the most populated class was selected, resulting in the final set of 166,619 particles.
初期モデル
モデルのタイプ: NONE 詳細: Starting model was generating using ab-initio procedure in cryoSPARC
最終 再構成
使用したクラス数: 1 / 想定した対称性 - 点群: C1 (非対称) / アルゴリズム: FOURIER SPACE / 解像度のタイプ: BY AUTHOR / 解像度: 4.31 Å / 解像度の算出法: FSC 0.143 CUT-OFF / ソフトウェア - 名称: cryoSPARC (ver. 2) / 詳細: Non Uniform refinement in cryoSPARC / 使用した粒子像数: 166619
初期 角度割当
タイプ: PROJECTION MATCHING
最終 角度割当
タイプ: PROJECTION MATCHING
最終 3次元分類
クラス数: 2 / ソフトウェア - 名称: RELION (ver. 3.1) 詳細: Particles were subjected to two rounds of 3D classification using ab-initio reconstruction in cryoSPARC-2 with 2 classes and class similarity set to 0. At the end of each round, the most ...詳細: Particles were subjected to two rounds of 3D classification using ab-initio reconstruction in cryoSPARC-2 with 2 classes and class similarity set to 0. At the end of each round, the most populated class was selected, resulting in the final set of 166,619 particles.
chain_id: F, source_name: PDB, initial_model_type: experimental model
詳細
The atomistic models of monomeric SARS-CoV-2 S1 protein and a Fab molecule, extracted from PDB entries 7A92 and 5WI9, were docked in the cryo-EM map using Chimera. The NTD and the RBD of the spike subunit were replaced with the crystal structures from PDB entries 7B62 and 7OAO, respectively. Guided by the cryo-EM map, the model was adjusted and extended interactively in Coot and refined using phenix.real_space_refine.
精密化
空間: REAL / プロトコル: OTHER / 温度因子: 81 / 当てはまり具合の基準: Correlation coefficient
得られたモデル
PDB-7zbu: CryoEM structure of SARS-CoV-2 spike monomer in complex with neutralising antibody P008_60
 ムービー
ムービー コントローラー
コントローラー


 データを開く
データを開く
 基本情報
基本情報
 マップデータ
マップデータ 試料
試料 キーワード
キーワード 機能・相同性情報
機能・相同性情報 Homo sapiens (ヒト) /
Homo sapiens (ヒト) / 
 Severe acute respiratory syndrome coronavirus 2 (ウイルス)
Severe acute respiratory syndrome coronavirus 2 (ウイルス) データ登録者
データ登録者 英国, 5件
英国, 5件  引用
引用 構造の表示
構造の表示
 ダウンロードとリンク
ダウンロードとリンク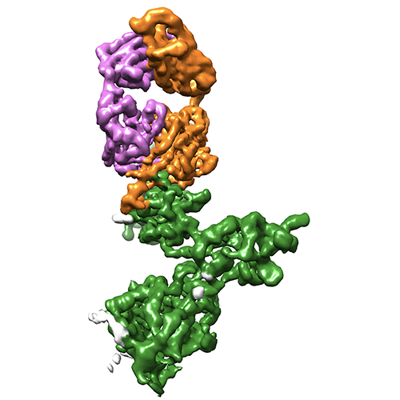 emd_14591.png
emd_14591.png http://ftp.pdbj.org/pub/emdb/structures/EMD-14591
http://ftp.pdbj.org/pub/emdb/structures/EMD-14591



 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)













































 試料の構成要素
試料の構成要素
 解析
解析 電子顕微鏡法
電子顕微鏡法 FIELD EMISSION GUN
FIELD EMISSION GUN




