 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-32571: Cryo-EM structure of GH31 alpha-1,3-glucosidase from Lactococcus ... -
+ Open data
Open data

- Basic information
Basic information
| Entry |  | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Cryo-EM structure of GH31 alpha-1,3-glucosidase from Lactococcus lactis subsp. cremoris | |||||||||
 Map data Map data | ||||||||||
 Sample Sample |
| |||||||||
| Function / homology |  Function and homology information Function and homology informationHydrolases; Glycosylases; Glycosidases, i.e. enzymes that hydrolyse O- and S-glycosyl compounds / hydrolase activity, hydrolyzing O-glycosyl compounds / carbohydrate metabolic process Similarity search - Function | |||||||||
| Biological species |  Lactococcus lactis subsp. cremoris MG1363 (lactic acid bacteria) Lactococcus lactis subsp. cremoris MG1363 (lactic acid bacteria) | |||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 2.73 Å | |||||||||
 Authors Authors | Ikegaya M / Moriya T / Adachi N / Kawasaki M / Park EY / Miyazaki T | |||||||||
| Funding support |  Japan, 1 items Japan, 1 items
| |||||||||
 Citation Citation | Journal: J Biol Chem / Year: 2022 Title: Structural basis of the strict specificity of a bacterial GH31 α-1,3-glucosidase for nigerooligosaccharides. Authors: Marina Ikegaya / Toshio Moriya / Naruhiko Adachi / Masato Kawasaki / Enoch Y Park / Takatsugu Miyazaki / Abstract: Carbohydrate-active enzymes are involved in the degradation, biosynthesis, and modification of carbohydrates and vary with the diversity of carbohydrates. The glycoside hydrolase (GH) family 31 is ...Carbohydrate-active enzymes are involved in the degradation, biosynthesis, and modification of carbohydrates and vary with the diversity of carbohydrates. The glycoside hydrolase (GH) family 31 is one of the most diverse families of carbohydrate-active enzymes, containing various enzymes that act on α-glycosides. However, the function of some GH31 groups remains unknown, as their enzymatic activity is difficult to estimate due to the low amino acid sequence similarity between characterized and uncharacterized members. Here, we performed a phylogenetic analysis and discovered a protein cluster (GH31_u1) sharing low sequence similarity with the reported GH31 enzymes. Within this cluster, we showed that a GH31_u1 protein from Lactococcus lactis (LlGH31_u1) and its fungal homolog demonstrated hydrolytic activities against nigerose [α-D-Glcp-(1→3)-D-Glc]. The k/K values of LlGH31_u1 against kojibiose and maltose were 13% and 2.1% of that against nigerose, indicating that LlGH31_u1 has a higher specificity to the α-1,3 linkage of nigerose than other characterized GH31 enzymes, including eukaryotic enzymes. Furthermore, the three-dimensional structures of LlGH31_u1 determined using X-ray crystallography and cryogenic electron microscopy revealed that LlGH31_u1 forms a hexamer and has a C-terminal domain comprising four α-helices, suggesting that it contributes to hexamerization. Finally, crystal structures in complex with nigerooligosaccharides and kojibiose along with mutational analysis revealed the active site residues involved in substrate recognition in this enzyme. This study reports the first structure of a bacterial GH31 α-1,3-glucosidase and provides new insight into the substrate specificity of GH31 enzymes and the physiological functions of bacterial and fungal GH31_u1 members. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Supplemental images |
|---|

- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_32571.map.gz | 478 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-32571-v30.xmlemd-32571.xml | 18.9 KB 18.9 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_32571_fsc.xml | 18.1 KB | Display | FSC data file |
| Images | 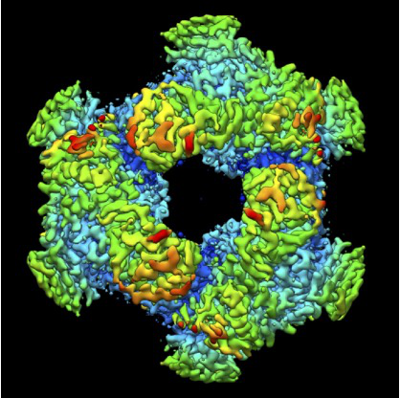 emd_32571.png emd_32571.png | 206.9 KB | ||
| Masks | emd_32571_msk_1.map | 512 MB | Mask map | |
| Others | emd_32571_half_map_1.map.gzemd_32571_half_map_2.map.gz | 412.4 MB 412.3 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-32571ftp://ftp.pdbj.org/pub/emdb/structures/EMD-32571 http://ftp.pdbj.org/pub/emdb/structures/EMD-32571ftp://ftp.pdbj.org/pub/emdb/structures/EMD-32571 | HTTPS FTP |
-Related structure data
| Related structure data |  7wlgMC  7wj9C  7wjaC  7wjbC  7wjcC  7wjdC  7wjeC  7wjfC M: atomic model generated by this map C: citing same article ( |
|---|---|
| Similar structure data | |
| EM raw data | EMPIAR-11171 (Title: Cryo-EM structure of GH31 alpha-1,3-glucosidase from Lactococcus lactis subsp. cremoris Data size: 911.2 Data #1: Cryo-EM structure of GH31 alpha-1,3-glucosidase from Lactococcus lactis subsp. cremoris [micrographs - multiframe]) |
-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_32571.map.gz / Format: CCP4 / Size: 512 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 0.88 Å | ||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
|
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















-Supplemental data
-Mask #1
| File | emd_32571_msk_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
| Density Histograms |








-Half map: #1
| File | emd_32571_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
| Density Histograms |






-Half map: #2
| File | emd_32571_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
| Density Histograms |








- Sample components
Sample components
-Entire : LlGH31_u1 hexamer
| Entire | Name: LlGH31_u1 hexamer |
|---|---|
| Components |
|
-Supramolecule #1: LlGH31_u1 hexamer
| Supramolecule | Name: LlGH31_u1 hexamer / type: complex / Chimera: Yes / ID: 1 / Parent: 0 / Macromolecule list: all / Details: homo hexamer |
|---|---|
| Source (natural) | Organism: Lactococcus lactis subsp. cremoris MG1363 (lactic acid bacteria) |
| Recombinant expression | Organism: |
| Molecular weight | Theoretical: 516 KDa |
-Macromolecule #1: LlGH31_u1
| Macromolecule | Name: LlGH31_u1 / type: protein_or_peptide / ID: 1 / Enantiomer: LEVO / EC number: ec: 3.2.1.84 |
|---|---|
| Source (natural) | Organism: Lactococcus lactis subsp. cremoris MG1363 (lactic acid bacteria) Strain: MG1363 |
| Recombinant expression | Organism: |
| Sequence | String: MGSSHHHHHH SSGLVPRGSH MVSELESKYM NNNIIKFDKA RFTVLTEHLI RIEYSETGEF EERMTQMVQN REFSEVNFDI IEKEETIEII TSTVHLYYNG GEFTNASLFA DVKFNFSVYS NRWYFGEKSD GNLKGTTRTL DMIDGECPLE DGIMSKNGFA VLADKGKVLT ...String: MGSSHHHHHH SSGLVPRGSH MVSELESKYM NNNIIKFDKA RFTVLTEHLI RIEYSETGEF EERMTQMVQN REFSEVNFDI IEKEETIEII TSTVHLYYNG GEFTNASLFA DVKFNFSVYS NRWYFGEKSD GNLKGTTRTL DMIDGECPLE DGIMSKNGFA VLADKGKVLT EVGDIAGNSV STIDLYLFAY GRDYRQALKD FYQLTGNTPK LPRFALGNWW SRYYDYSDKS YLALMDKFTD KKVPLSVSVI DMDWHKVSEV PSRFGSGWTG YSWNKKLFPN PENFIDELHQ RKLKVTLNDH PADGIRAFED PYPQVAQTLD LNTELEEAAK FDFDNLKFRK AYFEEVHGPL EKEGVDFWWI DWQQGAISKS GVDPLWLLNH YQYQNAQKKH KNNIILSRYA GPGSHRYPLG FSGDSVISWA SLDFQPYFTS TASNIGYTWW SHDIGGHMQG YKDAELSLRW LQFGVFSPIN RLHSSKSEFT SKEPWHFDAV IEQSMIDFLQ LRHQLIPYLY SANLITASEG RALVEPLYYE YPMEEEAYQH RNQYLFGEQL MVAPITEKMN SLLQMGSVEV WFPEGTWYDF FSGQPYDGKV SLKVYREITE MPVFAKAGAI IPLDKNPLKK EEIPSEIIWK IFPGADGEYL LLEEDNETKA EFVNGIFTVT SKKESSRKHT IIYGEHEIVS AKRGEFSIDL NGKEENFDWN FSTALFRRLD IAEISYEQKD EILQQLSLIE EHEKQVAFIK TNENQELQNS LFELLYSGK |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 0.58 mg/mL | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Buffer | pH: 7 Component:
| |||||||||
| Grid | Model: Quantifoil R1.2/1.3 / Material: COPPER / Mesh: 300 / Support film - Material: CARBON / Support film - topology: HOLEY / Pretreatment - Type: GLOW DISCHARGE / Pretreatment - Atmosphere: AIR / Details: The grid was washed by acetone prior to use. | |||||||||
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 100 % / Chamber temperature: 291 K / Instrument: FEI VITROBOT MARK IV / Details: Blotting time was 5 seconds (blot force 15). | |||||||||
| Details | This sample was mono-disperse. |
- Electron microscopy
Electron microscopy
| Microscope | TFS TALOS |
|---|---|
| Image recording | Film or detector model: FEI FALCON III (4k x 4k) / Detector mode: COUNTING / Digitization - Dimensions - Width: 4096 pixel / Digitization - Dimensions - Height: 4096 pixel / Number grids imaged: 1 / Number real images: 995 / Average exposure time: 65.95 sec. / Average electron dose: 50.0 e/Å2 |
| Electron beam | Acceleration voltage: 200 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | C2 aperture diameter: 50.0 µm / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2.7 mm / Nominal defocus max: 2.0 µm / Nominal defocus min: 0.8 µm / Nominal magnification: 120000 |
| Sample stage | Cooling holder cryogen: NITROGEN |

