National Institutes of Health/National Institute of General Medical Sciences (NIH/NIGMS)
GM123496
United States
Citation
Journal: Elife / Year: 2022 Title: Structure of SARS-CoV-2 M protein in lipid nanodiscs. Authors: Kimberly A Dolan / Mandira Dutta / David M Kern / Abhay Kotecha / Gregory A Voth / Stephen G Brohawn / Abstract: SARS-CoV-2 encodes four structural proteins incorporated into virions, spike (S), envelope (E), nucleocapsid (N), and membrane (M). M plays an essential role in viral assembly by organizing other ...SARS-CoV-2 encodes four structural proteins incorporated into virions, spike (S), envelope (E), nucleocapsid (N), and membrane (M). M plays an essential role in viral assembly by organizing other structural proteins through physical interactions and directing them to sites of viral budding. As the most abundant protein in the viral envelope and a target of patient antibodies, M is a compelling target for vaccines and therapeutics. Still, the structure of M and molecular basis for its role in virion formation are unknown. Here, we present the cryo-EM structure of SARS-CoV-2 M in lipid nanodiscs to 3.5 Å resolution. M forms a 50 kDa homodimer that is structurally related to the SARS-CoV-2 ORF3a viroporin, suggesting a shared ancestral origin. Structural comparisons reveal how intersubunit gaps create a small, enclosed pocket in M and large open cavity in ORF3a, consistent with a structural role and ion channel activity, respectively. M displays a strikingly electropositive cytosolic surface that may be important for interactions with N, S, and viral RNA. Molecular dynamics simulations show a high degree of structural rigidity in a simple lipid bilayer and support a role for M homodimers in scaffolding viral assembly. Together, these results provide insight into roles for M in coronavirus assembly and structure.
EMPIAR-11067 (Title: Structure of SARS-CoV-2 M protein in lipid nanodiscs / Data size: 4.0 TB Data #1: SARS-CoV-2 M protein in an MSP1E3D1 lipid nanodisc [micrographs - multiframe])
Model: Quantifoil R1.2/1.3 / Material: GOLD / Mesh: 300 / Support film - Material: GOLD / Support film - topology: HOLEY / Pretreatment - Type: GLOW DISCHARGE
Vitrification
Cryogen name: ETHANE / Chamber humidity: 100 % / Chamber temperature: 277 K / Instrument: FEI VITROBOT MARK IV / Details: blot force 1, wait time 5s, blot time 3s.
-
Electron microscopy
Microscope
FEI TITAN KRIOS
Image recording
Film or detector model: FEI FALCON IV (4k x 4k) / Average electron dose: 50.0 e/Å2
Electron beam
Acceleration voltage: 300 kV / Electron source: FIELD EMISSION GUN
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information
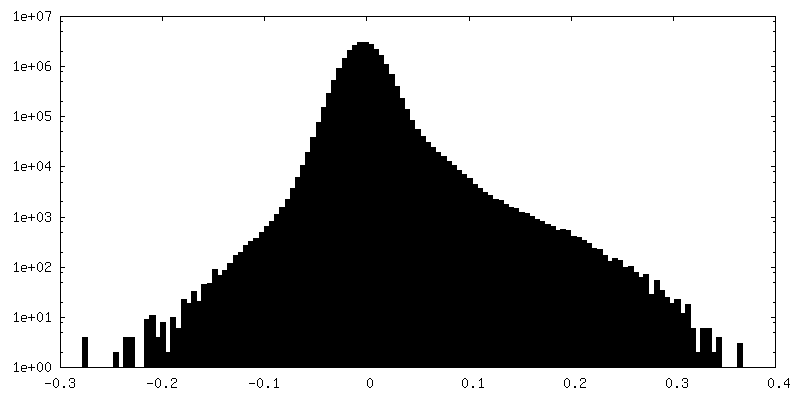
 Map data
Map data Sample
Sample Keywords
Keywords Function and homology information
Function and homology information
 Severe acute respiratory syndrome coronavirus 2
Severe acute respiratory syndrome coronavirus 2 Authors
Authors United States, 1 items
United States, 1 items  Citation
Citation
 Structure visualization
Structure visualization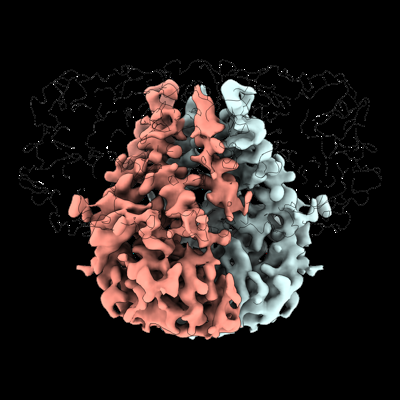
 Downloads & links
Downloads & links emd_26993.png
emd_26993.png http://ftp.pdbj.org/pub/emdb/structures/EMD-26993
http://ftp.pdbj.org/pub/emdb/structures/EMD-26993






 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)

























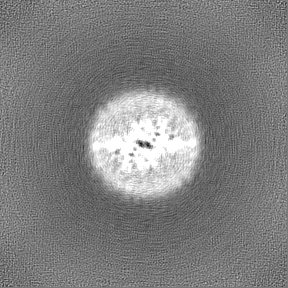
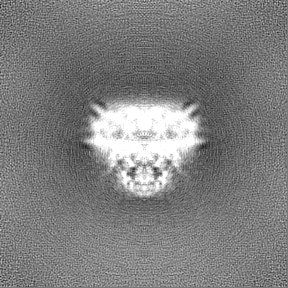
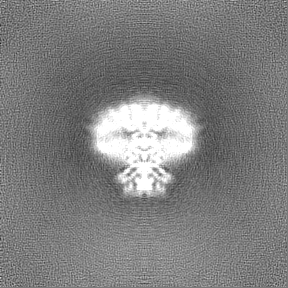



 Sample components
Sample components
 Spodoptera frugiperda (fall armyworm)
Spodoptera frugiperda (fall armyworm) Processing
Processing Electron microscopy
Electron microscopy FIELD EMISSION GUN
FIELD EMISSION GUN
