 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-26696: Cryo-EM structure of the S. cerevisiae chromatin remodeler Yta7 h... -
+ Open data
Open data

- Basic information
Basic information
| Entry |  | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Cryo-EM structure of the S. cerevisiae chromatin remodeler Yta7 hexamer bound to ATPgS and histone H3 tail in state II | |||||||||
 Map data Map data | Primary map that sharpen by deep enhancer | |||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords | AAA+ ATPase / chromatin remodeler / TRANSFERASE | |||||||||
| Function / homology |  Function and homology information Function and homology informationATP-dependent histone chaperone activity / positive regulation of invasive growth in response to glucose limitation / sexual sporulation resulting in formation of a cellular spore / cupric reductase (NADH) activity / global genome nucleotide-excision repair / RNA polymerase I upstream activating factor complex / Condensation of Prophase Chromosomes / : / : / : ...ATP-dependent histone chaperone activity / positive regulation of invasive growth in response to glucose limitation / sexual sporulation resulting in formation of a cellular spore / cupric reductase (NADH) activity / global genome nucleotide-excision repair / RNA polymerase I upstream activating factor complex / Condensation of Prophase Chromosomes / : / : / : / Assembly of the ORC complex at the origin of replication / CENP-A containing chromatin assembly / Oxidative Stress Induced Senescence / nucleosome disassembly / ATP-dependent chromatin remodeler activity / RNA Polymerase I Promoter Escape / positive regulation of transcription by RNA polymerase I / nucleolar large rRNA transcription by RNA polymerase I / Estrogen-dependent gene expression / rRNA transcription / intracellular copper ion homeostasis / chromosome, centromeric region / Hydrolases; Acting on acid anhydrides; In phosphorus-containing anhydrides / CENP-A containing nucleosome / transcription initiation-coupled chromatin remodeling / aerobic respiration / DNA-templated DNA replication / structural constituent of chromatin / nucleosome / nucleosome assembly / chromosome / chromatin organization / histone binding / chromatin remodeling / protein heterodimerization activity / chromatin binding / regulation of DNA-templated transcription / chromatin / negative regulation of transcription by RNA polymerase II / ATP hydrolysis activity / positive regulation of transcription by RNA polymerase II / DNA binding / ATP binding / nucleus / cytosol Similarity search - Function | |||||||||
| Biological species |  | |||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 3.0 Å | |||||||||
 Authors Authors | Wang F / Feng X / Li H | |||||||||
| Funding support |  United States, 1 items United States, 1 items
| |||||||||
 Citation Citation | Journal: J Biol Chem / Year: 2023 Title: The Saccharomyces cerevisiae Yta7 ATPase hexamer contains a unique bromodomain tier that functions in nucleosome disassembly. Authors: Feng Wang / Xiang Feng / Qing He / Hua Li / Huilin Li / Abstract: The Saccharomyces cerevisiae Yta7 is a chromatin remodeler harboring a histone-interacting bromodomain (BRD) and two AAA+ modules. It is not well understood how Yta7 recognizes the histone H3 tail to ...The Saccharomyces cerevisiae Yta7 is a chromatin remodeler harboring a histone-interacting bromodomain (BRD) and two AAA+ modules. It is not well understood how Yta7 recognizes the histone H3 tail to promote nucleosome disassembly for DNA replication or RNA transcription. By cryo-EM analysis, here we show that Yta7 assembles a three-tiered hexamer with a top BRD tier, a middle AAA1 tier, and a bottom AAA2 tier. Unexpectedly, the Yta7 BRD stabilizes a four-stranded β-helix, termed BRD-interacting motif (BIM), of the largely disordered N-terminal region. The BIM motif is unique to the baker's yeast, and we show both BRD and BIM contribute to nucleosome recognition. We found that Yta7 binds both acetylated and nonacetylated H3 peptides but with a higher affinity for the unmodified peptide. This property is consistent with the absence of key residues of canonical BRDs involved in acetylated peptide recognition and the role of Yta7 in general nucleosome remodeling. Interestingly, the BRD tier exists in a spiral and a flat-ring form on top of the Yta7 AAA+ hexamer. The spiral is likely in a nucleosome-searching mode because the bottom BRD blocks the entry to the AAA+ chamber. The flat ring may be in a nucleosome disassembly state because the entry is unblocked and the H3 peptide has entered the AAA+ chamber and is stabilized by the AAA1 pore loops 1 and 2. Indeed, we show that the BRD tier is a flat ring when bound to the nucleosome. Overall, our study sheds light on the nucleosome disassembly by Yta7. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Supplemental images |
|---|

- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_26696.map.gz | 215.5 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-26696-v30.xmlemd-26696.xml | 25.8 KB 25.8 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_26696_fsc.xml | 14.2 KB | Display | FSC data file |
| Images | 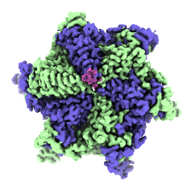 emd_26696.png emd_26696.png | 45 KB | ||
| Filedesc metadata | emd-26696.cif.gz | 7.3 KB | ||
| Others | emd_26696_additional_1.map.gzemd_26696_additional_2.map.gzemd_26696_half_map_1.map.gzemd_26696_half_map_2.map.gz | 193.2 MB 228.5 MB 194.1 MB 194.2 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-26696ftp://ftp.pdbj.org/pub/emdb/structures/EMD-26696 http://ftp.pdbj.org/pub/emdb/structures/EMD-26696ftp://ftp.pdbj.org/pub/emdb/structures/EMD-26696 | HTTPS FTP |
-Related structure data
| Related structure data |  7uqjMC  7uqiC  7uqkC C: citing same article ( M: atomic model generated by this map |
|---|---|
| Similar structure data |




-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| Related items in Molecule of the Month |









-Map
| File | Download / File: emd_26696.map.gz / Format: CCP4 / Size: 244.1 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Primary map that sharpen by deep enhancer | ||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 0.828 Å | ||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
|
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















-Supplemental data
-Additional map: Raw map
| File | emd_26696_additional_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Raw map | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |








-Additional map: Relion sharpen map
| File | emd_26696_additional_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Relion sharpen map | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |








-Half map: #2
| File | emd_26696_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
| Density Histograms |








-Half map: #1
| File | emd_26696_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
| Density Histograms |






- Sample components
Sample components
-Entire : Complex of Yta7 with H3N tail
| Entire | Name: Complex of Yta7 with H3N tail |
|---|---|
| Components |
|
-Supramolecule #1: Complex of Yta7 with H3N tail
| Supramolecule | Name: Complex of Yta7 with H3N tail / type: complex / ID: 1 / Parent: 0 / Macromolecule list: #1-#2 Details: The translocation state of Histone 3 N-terminal peptide by Yta7 |
|---|---|
| Source (natural) | Organism: |
| Molecular weight | Theoretical: 930 KDa |
-Macromolecule #1: ATPase histone chaperone YTA7
| Macromolecule | Name: ATPase histone chaperone YTA7 / type: protein_or_peptide / ID: 1 / Number of copies: 6 / Enantiomer: LEVO EC number: Hydrolases; Acting on acid anhydrides; In phosphorus-containing anhydrides |
|---|---|
| Source (natural) | Organism: |
| Molecular weight | Theoretical: 162.182969 KDa |
| Recombinant expression | Organism: |
| Sequence | String: HHHHHHHHHH TSGSMDYKDH DGDYKDHDID YKDDDDKMAR NLRNRRGSDV EDASNAKVGY ETQIKDENGI IHTTTRSLRK INYAEIEKV FDFLEDDQVM DKDETPVDVT SDEHHNNNQK GDDEDDDVDL VSPHENARTN EELTNERNLR KRKAHDPEED D ESFHEEDV ...String: HHHHHHHHHH TSGSMDYKDH DGDYKDHDID YKDDDDKMAR NLRNRRGSDV EDASNAKVGY ETQIKDENGI IHTTTRSLRK INYAEIEKV FDFLEDDQVM DKDETPVDVT SDEHHNNNQK GDDEDDDVDL VSPHENARTN EELTNERNLR KRKAHDPEED D ESFHEEDV DDDEEEEEAD EFEDEYLDED SKDNNRRRRA ADRKFVVPDP DDDEEYDEDD EEGDRISHSA SSKRLKRANS RR TRSSRHP ETPPPVRRAL RSRTRHSRTS NEENDDENDN SRNEALTLAD EIRELQEDSP IREKRFLRER TKPVNYKLPP PLT ASNAEE FIDKNNNALS FHNPSPARRG RGGWNASQNS GPTRRLFPTG GPFGGNDVTT IFGKNTNFYN QVPSAFSDNN NNKL ILDSD SSDDEILPLG VTPKTKKENT QKKKKKKPEI ADLDPLGVDM NVNFDDIGGL DNYIDQLKEM VALPLLYPEL YQNFN ITPP RGVLFHGPPG TGKTLMARAL AASCSSDERK ITFFMRKGAD ILSKWVGEAE RQLRLLFEEA KKHQPSIIFF DEIDGL APV RSSKQEQIHA SIVSTLLALM DGMDNRGQVI VIGATNRPDA VDPALRRPGR FDREFYFPLP DVKARFKILQ IQTRKWS SP LSTNFIDKLA FLTKGYGGAD LRSLCTEAAL ISIQRSFPQI YRSNDKLLVD PSKIKVKVSD FMLALKKIVP SSARSTGS S PQPLPELIKP LLADQLNNLK NKLDYMLNIK DTTFQRNTSL LQNFIDYEEY SGEEEEHDKY GGNEDTSSFR SYEFFESMA ESQICKPRLL INGPKGNGQQ YVGAAILNYL EEFNVQNLDL ASLVSESSRT IEAAVVQSFM EAKKRQPSVV FIPNLDIWIN TIPENVILV LSGLFRSLQS NEKILLLCLA ENLDISEVKN GILSDFAFDK NIFQLHKPSK ENITRYFSNL IELLKTKPSD I PMKKRRVK PLPELQKVTS NAAPTNFDEN GEPLSEKVVL RRKLKSFQHQ DMRLKNVLKI KLSGLMDLFK NRYKRFRKPP ID DAFLVHL FEPETSNDPN WQPAYIKDEN MILEVSTGRK FFNMDLDIVE ERLWNGYYSE PKQFLKDIEL IYRDANTIGD RER VIKASE MFANAQMGIE EISTPDFIQE CKATRQRDLE RQELFLEDEE KRAAMELEAK EQSQENILQE PDLKDNKANE FGVA AGNQL QAQLQTTINT ASIVNNSEVP QPIDTNLYKK EIPAAIPSAV DKEKAVIPED SGANEEYTTE LIQATCTSEI TTDDD ERAR KEPKENEDSL QTQVTEENFS KIDANTNNIN HVKEIQSVNK PNSLHETVEK RERSPIPKEV VEPEQGKKSD KELILT PEQ IKKVSACLIE HCQNFTVSQL EDVHSSVAKI IWKSKSAWDK TGTVDEIIKF LSE UniProtKB: ATPase histone chaperone YTA7 |
-Macromolecule #2: Histone H3
| Macromolecule | Name: Histone H3 / type: protein_or_peptide / ID: 2 / Number of copies: 1 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: |
| Molecular weight | Theoretical: 2.680181 KDa |
| Sequence | String: MARTKQTARK STGGKAPRKQ LASKA UniProtKB: Histone H3 |
-Macromolecule #3: ADENOSINE-5'-DIPHOSPHATE
| Macromolecule | Name: ADENOSINE-5'-DIPHOSPHATE / type: ligand / ID: 3 / Number of copies: 1 / Formula: ADP |
|---|---|
| Molecular weight | Theoretical: 427.201 Da |
| Chemical component information |  ChemComp-ADP: |
-Macromolecule #4: PHOSPHOTHIOPHOSPHORIC ACID-ADENYLATE ESTER
| Macromolecule | Name: PHOSPHOTHIOPHOSPHORIC ACID-ADENYLATE ESTER / type: ligand / ID: 4 / Number of copies: 5 / Formula: AGS |
|---|---|
| Molecular weight | Theoretical: 523.247 Da |
| Chemical component information |  ChemComp-AGS: |
-Macromolecule #5: MAGNESIUM ION
| Macromolecule | Name: MAGNESIUM ION / type: ligand / ID: 5 / Number of copies: 4 / Formula: MG |
|---|---|
| Molecular weight | Theoretical: 24.305 Da |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 10 mg/mL | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Buffer | pH: 7.6 Component:
Details: Solution was made fresh and detergent was added to solve preference orientation issue. | ||||||||||||||||||||||||
| Grid | Model: Quantifoil R2/1 / Material: COPPER / Mesh: 300 / Support film - Material: GOLD / Support film - topology: CONTINUOUS / Pretreatment - Type: GLOW DISCHARGE / Pretreatment - Time: 30 sec. / Pretreatment - Atmosphere: AIR / Pretreatment - Pressure: 101.325 kPa / Details: The grids were doulbe blots | ||||||||||||||||||||||||
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 100 % / Chamber temperature: 281 K / Instrument: FEI VITROBOT MARK IV / Details: Blot 3S, blot forth 3. | ||||||||||||||||||||||||
| Details | The sample was a novel chromatin remodeler and a AAA+ ATPase. |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Temperature | Min: 193.0 K / Max: 193.0 K |
| Image recording | Film or detector model: GATAN K3 (6k x 4k) / Average electron dose: 65.0 e/Å2 Details: A total of 75 frames were recorded for each micrograph stack. |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | C2 aperture diameter: 70.0 µm / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2.7 mm / Nominal defocus max: 2.0 µm / Nominal defocus min: 1.0 µm / Nominal magnification: 105000 |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER / Cooling holder cryogen: NITROGEN |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
+Image processing


-Atomic model buiding 1
| Refinement | Space: REAL / Protocol: RIGID BODY FIT / Target criteria: Correlation coefficient |
|---|---|
| Output model | PDB-7uqj: |
