 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry |  | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
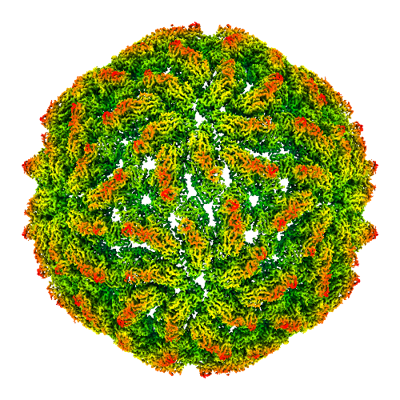
| Title | Empty capsid of Saccharomyces cerevisiae virus L-BCLa | |||||||||
 Map data Map data | ||||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords | capsid protein dimer / icosahedral asymmetric unit / totivirus capsid / VIRUS | |||||||||
| Function / homology | Major coat protein, L-A virus / L-A virus major coat protein superfamily / L-A virus, major coat protein / viral capsid / viral translational frameshifting / RNA binding / Major capsid protein Function and homology information Function and homology information | |||||||||
| Biological species |  Saccharomyces cerevisiae virus L-BC (La) Saccharomyces cerevisiae virus L-BC (La) | |||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 2.9 Å | |||||||||
 Authors Authors | Grybchuk D / Prochazkova M | |||||||||
| Funding support |  Czech Republic, 1 items Czech Republic, 1 items
| |||||||||
 Citation Citation | Journal: Commun Biol / Year: 2022 Title: Structures of L-BC virus and its open particle provide insight into Totivirus capsid assembly. Authors: Danyil Grybchuk / Michaela Procházková / Tibor Füzik / Aleksandras Konovalovas / Saulius Serva / Vyacheslav Yurchenko / Pavel Plevka / Abstract: L-BC virus persists in the budding yeast Saccharomyces cerevisiae, whereas other viruses from the family Totiviridae infect a diverse group of organisms including protists, fungi, arthropods, and ...L-BC virus persists in the budding yeast Saccharomyces cerevisiae, whereas other viruses from the family Totiviridae infect a diverse group of organisms including protists, fungi, arthropods, and vertebrates. The presence of totiviruses alters the fitness of the host organisms, for example, by maintaining the killer system in yeast or increasing the virulence of Leishmania guyanensis. Despite the importance of totiviruses for their host survival, there is limited information about Totivirus structure and assembly. Here we used cryo-electron microscopy to determine the structure of L-BC virus to a resolution of 2.9 Å. The L-BC capsid is organized with icosahedral symmetry, with each asymmetric unit composed of two copies of the capsid protein. Decamers of capsid proteins are stabilized by domain swapping of the C-termini of subunits located around icosahedral fivefold axes. We show that capsids of 9% of particles in a purified L-BC sample were open and lacked one decamer of capsid proteins. The existence of the open particles together with domain swapping within a decamer provides evidence that Totiviridae capsids assemble from the decamers of capsid proteins. Furthermore, the open particles may be assembly intermediates that are prepared for the incorporation of the virus (+) strand RNA. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Supplemental images |
|---|
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_14194.map.gz | 95.8 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-14194-v30.xmlemd-14194.xml | 15.3 KB 15.3 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_14194_fsc.xml | 19.7 KB | Display | FSC data file |
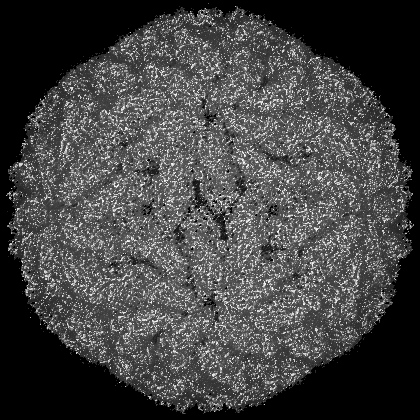
| Images |  emd_14194.png emd_14194.png | 226.8 KB | ||
| Filedesc metadata | emd-14194.cif.gz | 6.3 KB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-14194ftp://ftp.pdbj.org/pub/emdb/structures/EMD-14194 http://ftp.pdbj.org/pub/emdb/structures/EMD-14194ftp://ftp.pdbj.org/pub/emdb/structures/EMD-14194 | HTTPS FTP |
-Related structure data
| Related structure data |  7qwxMC  7qwzC  7ztsC  7zufC M: atomic model generated by this map C: citing same article ( |
|---|---|
| Similar structure data |



-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_14194.map.gz / Format: CCP4 / Size: 282.6 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.079 Å | ||||||||||||||||||||||||||||||||||||
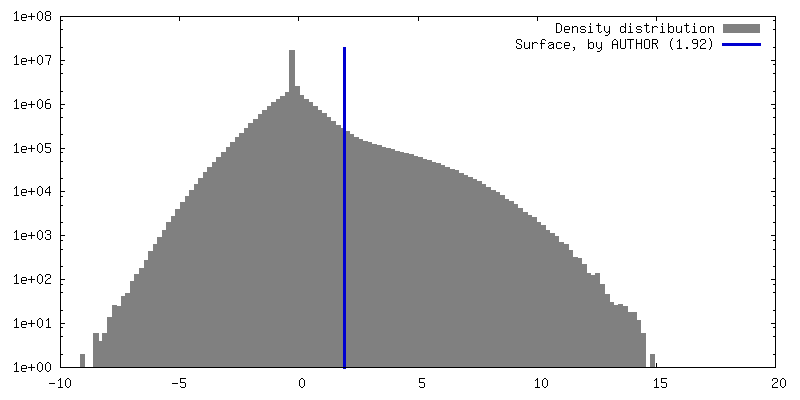
| Density |
| ||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
|
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)



















-Supplemental data
- Sample components
Sample components
-Entire : Saccharomyces cerevisiae virus L-BC (La)
| Entire | Name: Saccharomyces cerevisiae virus L-BC (La) |
|---|---|
| Components |
|
-Supramolecule #1: Saccharomyces cerevisiae virus L-BC (La)
| Supramolecule | Name: Saccharomyces cerevisiae virus L-BC (La) / type: virus / ID: 1 / Parent: 0 / Macromolecule list: all / NCBI-ID: 42478 / Sci species name: Saccharomyces cerevisiae virus L-BC (La) / Virus type: VIRION / Virus isolate: SPECIES / Virus enveloped: No / Virus empty: Yes |
|---|---|
| Host (natural) | Organism:  |
| Virus shell | Shell ID: 1 / Diameter: 400.0 Å / T number (triangulation number): 2 |
-Macromolecule #1: Major capsid protein
| Macromolecule | Name: Major capsid protein / type: protein_or_peptide / ID: 1 / Number of copies: 2 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: Saccharomyces cerevisiae virus L-BC (La) |
| Molecular weight | Theoretical: 78.393812 KDa |
| Sequence | String: MSSLLNSLLP EYFKPKTNLN INSSRVQYGF NARIDMQYED DSGTRKGSRP NAFMSNTVAF IGNYEGIIVD DIPILDGLRA DIFDTHGDL DMGLVEDALS KSTMIRRNVP TYTAYASELL YKRNLTSLFY NMLRLYYIKK WGSIKYEKDA IFYDNGHACL L NRQLFPKS ...String: MSSLLNSLLP EYFKPKTNLN INSSRVQYGF NARIDMQYED DSGTRKGSRP NAFMSNTVAF IGNYEGIIVD DIPILDGLRA DIFDTHGDL DMGLVEDALS KSTMIRRNVP TYTAYASELL YKRNLTSLFY NMLRLYYIKK WGSIKYEKDA IFYDNGHACL L NRQLFPKS RDASLESSLS LPEAEIAMLD PGLEFPEEDV PAILWHGRVS SRATCILGQA CSEFAPLAPF SIAHYSPQLT RK LFVNAPA GIEPSSGRYT HEDVKDAITI LVSANQAYTD FEAAYLMLAQ TLVSPVPRTA EASAWFINAG MVNMPTLSCA NGY YPALTN VNPYHRLDTW KDTLNHWVAY PDMLFYHSVA MIESCYVELG NVARVSDSDA INKYTFTELS VQGRPVMNRG IIVD LTLVA MRTGREISLP YPVSCGLTRT DALLQGTEIH VPVVVKDIDM PQYYNAIDKD VIEGQETVIK VKQLPPAMYP IYTYG INTT EFYSDHFEDQ VQVEMAPIDN GKAVFNDARK FSKFMSIMRM MGNDVTATDL VTGRKVSNWA DNSSGRFLYT DVKYEG QTA FLVDMDTVKA RDHCWVSIVD PNGTMNLSYK MTNFRAAMFS RNKPLYMTGG SVRTIATGNY RDAAERLRAM DETLRLK PF KITEKLDFRV AAYAIPSLSG SNMPSLHHQE QLQISEVDAE PINPIGEDEL PPDIE UniProtKB: Major capsid protein |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 1.5 mg/mL |
|---|---|
| Buffer | pH: 7.5 / Details: 20 mM Tris-HCl pH 7.5, 50 mM KCl, 10 mM MgCl2 |
| Grid | Model: Quantifoil R2/2 / Material: COPPER / Mesh: 200 / Support film - Material: CARBON / Support film - topology: HOLEY ARRAY / Pretreatment - Type: GLOW DISCHARGE / Pretreatment - Time: 15 sec. / Pretreatment - Atmosphere: OTHER / Pretreatment - Pressure: 0.007 kPa / Details: Glow discharge current 7 mA |
| Vitrification | Cryogen name: ETHANE-PROPANE / Chamber humidity: 100 % / Chamber temperature: 277 K / Instrument: FEI VITROBOT MARK IV / Details: blot force 0, blot time 3 s, 4 C, 100% humidity. |
| Details | Virus was isolated by shearing with glass beads and overnight precipitation in 5% PEG-4000 and 500 mM NaCl |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Temperature | Min: 70.0 K / Max: 80.0 K |
| Specialist optics | Energy filter - Name: GIF Bioquantum / Energy filter - Slit width: 10 eV |
| Image recording | Film or detector model: GATAN K2 SUMMIT (4k x 4k) / Detector mode: COUNTING / Digitization - Dimensions - Width: 3600 pixel / Digitization - Dimensions - Height: 3600 pixel / Digitization - Frames/image: 1-30 / Number grids imaged: 1 / Number real images: 11977 / Average exposure time: 6.0 sec. / Average electron dose: 36.0 e/Å2 |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | C2 aperture diameter: 30.0 µm / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2.7 mm / Nominal defocus max: 1.7 µm / Nominal defocus min: 0.7000000000000001 µm / Nominal magnification: 130000 |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER / Cooling holder cryogen: NITROGEN |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
+Image processing
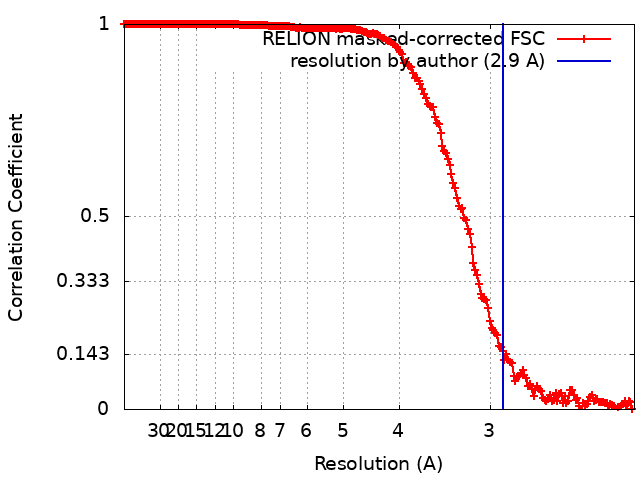
-Atomic model buiding 1
| Details | Initial model generated by RaptorX server |
|---|---|
| Refinement | Space: REAL / Protocol: FLEXIBLE FIT |
| Output model | PDB-7qwx: |
