 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-13802: Structure of full-length, dimeric, soluble somatic angiotensin I-... -
+ Open data
Open data

- Basic information
Basic information
| Entry |  | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Structure of full-length, dimeric, soluble somatic angiotensin I-converting enzyme | |||||||||
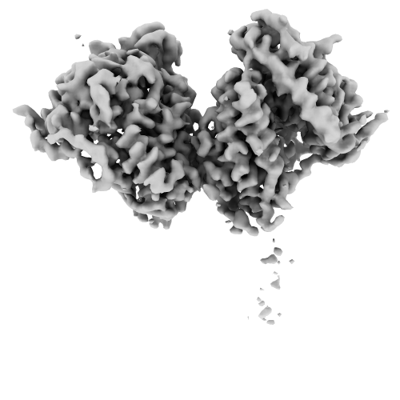
 Map data Map data | Globally-sharpened map of full-length dimeric somatic angiotensin I-converting enzyme showing interacting N-domains and disordered C-domains | |||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords | Zinc metalloprotease Dicarboxypeptidase Glycoprotein / HYDROLASE | |||||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | |||||||||
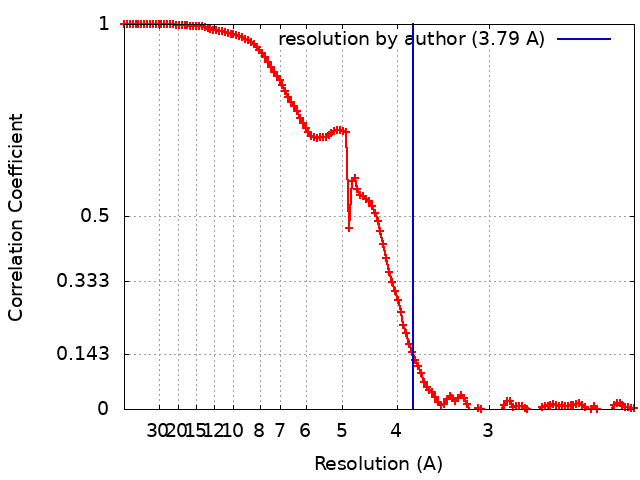
| Method | single particle reconstruction / cryo EM / Resolution: 3.79 Å | |||||||||
 Authors Authors | Lubbe L / Sewell BT / Sturrock ED | |||||||||
| Funding support |  United Kingdom, 1 items United Kingdom, 1 items
| |||||||||
 Citation Citation | Journal: EMBO J / Year: 2022 Title: Cryo-EM reveals mechanisms of angiotensin I-converting enzyme allostery and dimerization. Authors: Lizelle Lubbe / Bryan Trevor Sewell / Jeremy D Woodward / Edward D Sturrock /  Abstract: Hypertension (high blood pressure) is a major risk factor for cardiovascular disease, which is the leading cause of death worldwide. The somatic isoform of angiotensin I-converting enzyme (sACE) ...Hypertension (high blood pressure) is a major risk factor for cardiovascular disease, which is the leading cause of death worldwide. The somatic isoform of angiotensin I-converting enzyme (sACE) plays a critical role in blood pressure regulation, and ACE inhibitors are thus widely used to treat hypertension and cardiovascular disease. Our current understanding of sACE structure, dynamics, function, and inhibition has been limited because truncated, minimally glycosylated forms of sACE are typically used for X-ray crystallography and molecular dynamics simulations. Here, we report the first cryo-EM structures of full-length, glycosylated, soluble sACE (sACE ). Both monomeric and dimeric forms of the highly flexible apo enzyme were reconstructed from a single dataset. The N- and C-terminal domains of monomeric sACE were resolved at 3.7 and 4.1 Å, respectively, while the interacting N-terminal domains responsible for dimer formation were resolved at 3.8 Å. Mechanisms are proposed for intradomain hinging, cooperativity, and homodimerization. Furthermore, the observation that both domains were in the open conformation has implications for the design of sACE modulators. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Supplemental images |
|---|
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_13802.map.gz | 168.3 MB |  EMDB map data format EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-13802-v30.xmlemd-13802.xml | 23.5 KB 23.5 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_13802_fsc.xml | 12.5 KB | Display | FSC data file |
| Images |  emd_13802.png emd_13802.png | 63.8 KB | ||
| Masks | emd_13802_msk_1.mapemd_13802_msk_2.map | 178 MB 178 MB | Mask map | |
| Filedesc metadata | emd-13802.cif.gz | 6.7 KB | ||
| Others | emd_13802_additional_1.map.gzemd_13802_half_map_1.map.gzemd_13802_half_map_2.map.gz | 164.8 MB 165.4 MB 165.4 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-13802ftp://ftp.pdbj.org/pub/emdb/structures/EMD-13802 http://ftp.pdbj.org/pub/emdb/structures/EMD-13802ftp://ftp.pdbj.org/pub/emdb/structures/EMD-13802 | HTTPS FTP |
-Related structure data
| Related structure data |  7q3yC  7q49C  7q4cC  7q4dC  7q4eC C: citing same article ( |
|---|---|
| EM raw data | EMPIAR-10980 (Title: Cryo-EM structures of monomeric and dimeric human somatic angiotensin I-converting enzyme (apo form) Data size: 3.8 TB Data #1: Unaligned multi-frame cryo-EM micrographs of human somatic angiotensin I-converting enzyme in the apo state [micrographs - multiframe]) |





-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_13802.map.gz / Format: CCP4 / Size: 178 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Globally-sharpened map of full-length dimeric somatic angiotensin I-converting enzyme showing interacting N-domains and disordered C-domains | ||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.06 Å | ||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
|
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)




















-Supplemental data
-Mask #1
| File | emd_13802_msk_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
| Density Histograms |






-Mask #2
| File | emd_13802_msk_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
| Density Histograms |







-Additional map: Raw, unfiltered full map from non-uniform refinement of...
| File | emd_13802_additional_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Raw, unfiltered full map from non-uniform refinement of full-length dimeric somatic angiotensin I-converting enzyme | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |




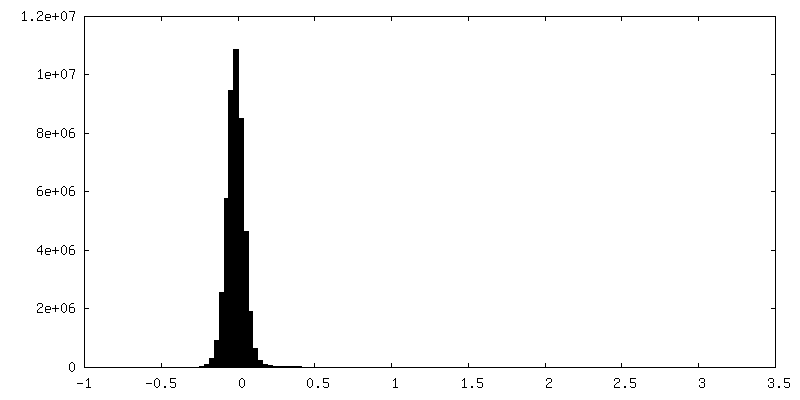
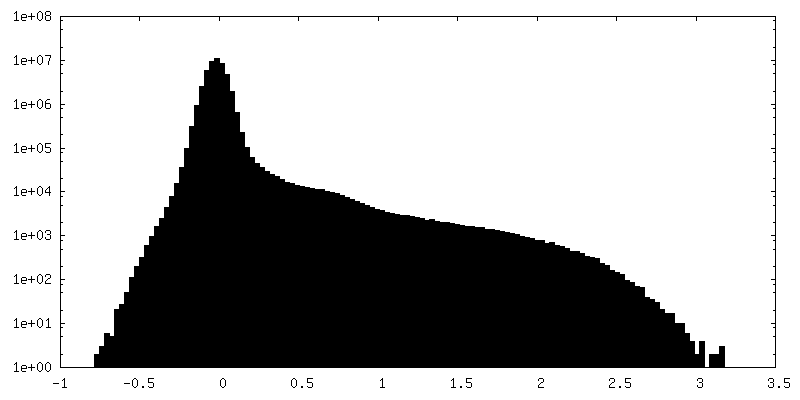
-Half map: Raw, unfiltered half-map A of full-length dimeric somatic...
| File | emd_13802_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Raw, unfiltered half-map A of full-length dimeric somatic angiotensin I-converting enzyme | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |





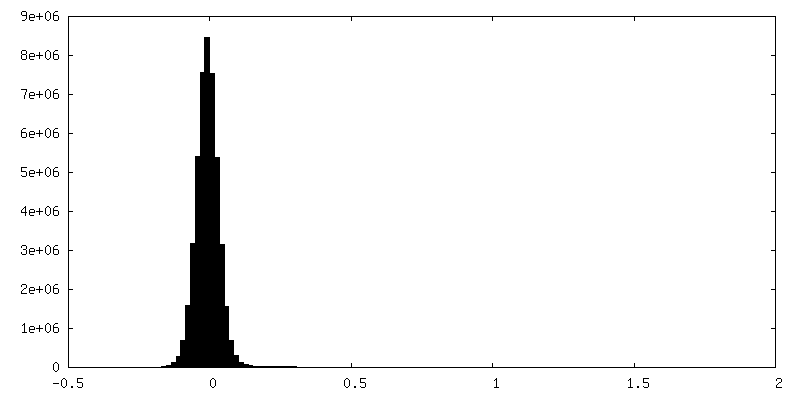
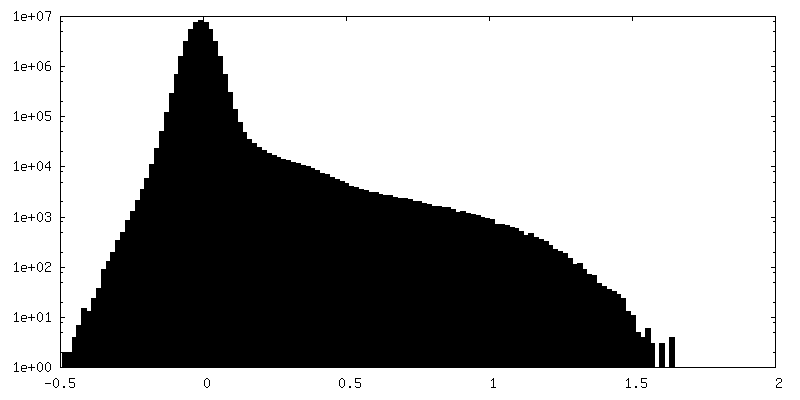
-Half map: Raw, unfiltered half-map B of full-length dimeric somatic...
| File | emd_13802_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Raw, unfiltered half-map B of full-length dimeric somatic angiotensin I-converting enzyme | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |



- Sample components
Sample components
-Entire : Full-length, soluble, dimeric somatic angiotensin I-converting enzyme
| Entire | Name: Full-length, soluble, dimeric somatic angiotensin I-converting enzyme |
|---|---|
| Components |
|
-Supramolecule #1: Full-length, soluble, dimeric somatic angiotensin I-converting enzyme
| Supramolecule | Name: Full-length, soluble, dimeric somatic angiotensin I-converting enzyme type: complex / ID: 1 / Parent: 0 / Macromolecule list: all |
|---|---|
| Source (natural) | Organism: Homo sapiens (human) |
| Molecular weight | Theoretical: 279 KDa |
-Macromolecule #1: Angiotensin I-converting enzyme
| Macromolecule | Name: Angiotensin I-converting enzyme / type: protein_or_peptide / ID: 1 Details: Soluble secreted form of human somatic angiotensin I-converting enzyme terminating at Ser1211 Enantiomer: LEVO / EC number: peptidyl-dipeptidase A |
|---|---|
| Source (natural) | Organism: Homo sapiens (human) |
| Recombinant expression | Organism:   Cricetulus griseus (Chinese hamster) Cricetulus griseus (Chinese hamster) |
| Sequence | String: LDPGLQPGNF SADEAGAQLF AQSYNSSAEQ V LFQSVAAS WAHDTNITAE NARRQEEAAL LSQEFAEAWG QKAKELYEPI WQNFTDPQLR RI IGAVRTL GSANLPLAKR QQYNALLSNM SRIYSTAKVC LPNKTATCWS LDPDLTNILA SSR SYAMLL FAWEGWHNAA ...String: LDPGLQPGNF SADEAGAQLF AQSYNSSAEQ V LFQSVAAS WAHDTNITAE NARRQEEAAL LSQEFAEAWG QKAKELYEPI WQNFTDPQLR RI IGAVRTL GSANLPLAKR QQYNALLSNM SRIYSTAKVC LPNKTATCWS LDPDLTNILA SSR SYAMLL FAWEGWHNAA GIPLKPLYED FTALSNEAYK QDGFTDTGAY WRSWYNSPTF EDDL EHLYQ QLEPLYLNLH AFVRRALHRR YGDRYINLRG PIPAHLLGDM WAQSWENIYD MVVPF PDKP NLDVTSTMLQ QGWNATHMFR VAEEFFTSLE LSPMPPEFWE GSMLEKPADG REVVCH ASA WDFYNRKDFR IKQCTRVTMD QLSTVHHEMG HIQYYLQYKD LPVSLRRGAN PGFHEAI GD VLALSVSTPE HLHKIGLLDR VTNDTESDIN YLLKMALEKI AFLPFGYLVD QWRWGVFS G RTPPSRYNFD WWYLRTKYQG ICPPVTRNET HFDAGAKFHV PNVTPYIRYF VSFVLQFQF HEALCKEAGY EGPLHQCDIY RSTKAGAKLR KVLQAGSSRP WQEVLKDMVG LDALDAQPLL KYFQLVTQW LQEQNQQNGE VLGWPEYQWH PPLPDNYPEG IDLVTDEAEA SKFVEEYDRT S QVVWNEYA EANWNYNTNI TTETSKILLQ KNMQIANHTL KYGTQARKFD VNQLQNTTIK RI IKKVQDL ERAALPAQEL EEYNKILLDM ETTYSVATVC HPNGSCLQLE PDLTNVMATS RKY EDLLWA WEGWRDKAGR AILQFYPKYV ELINQAARLN GYVDAGDSWR SMYETPSLEQ DLER LFQEL QPLYLNLHAY VRRALHRHYG AQHINLEGPI PAHLLGNMWA QTWSNIYDLV VPFPS APSM DTTEAMLKQG WTPRRMFKEA DDFFTSLGLL PVPPEFWNKS MLEKPTDGRE VVCHAS AWD FYNGKDFRIK QCTTVNLEDL VVAHHEMGHI QYFMQYKDLP VALREGANPG FHEAIGD VL ALSVSTPKHL HSLNLLSSEG GSDEHDINFL MKMALDKIAF IPFSYLVDQW RWRVFDGS I TKENYNQEWW SLRLKYQGLC PPVPRTQGDF DPGAKFHIPS SVPYIRYFVS FIIQFQFHE ALCQAAGHTG PLHKCDIYQS KEAGQRLATA MKLGFSRPWP EAMQLITGQP NMSASAMLSY FKPLLDWLR TENELHGEKL GWPQYNWTPN SARSEGPLPD S |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 1.5 mg/mL | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Buffer | pH: 7.5 Component:
Details: Solutions were prepared with deionized water | ||||||||||||
| Grid | Model: Quantifoil R2/2 / Material: COPPER / Mesh: 200 / Pretreatment - Type: GLOW DISCHARGE / Pretreatment - Time: 30 sec. | ||||||||||||
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 100 % / Chamber temperature: 277 K / Instrument: FEI VITROBOT MARK IV Details: Diluted protein (in buffer containing zinc chloride and sodium chloride) was incubated on ice for 30 minutes after which it was applied to the grid, incubated for 30 seconds, and blotted for ...Details: Diluted protein (in buffer containing zinc chloride and sodium chloride) was incubated on ice for 30 minutes after which it was applied to the grid, incubated for 30 seconds, and blotted for 3 seconds before plunging. | ||||||||||||
| Details | The protein was stored at 3.0mg/ml in 50mM HEPES (pH 7.5) and diluted immediately prior to grid preparation |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Image recording | Film or detector model: GATAN K3 (6k x 4k) / Digitization - Dimensions - Width: 5760 pixel / Digitization - Dimensions - Height: 4092 pixel / Number grids imaged: 1 / Number real images: 11628 / Average exposure time: 3.0 sec. / Average electron dose: 43.0 e/Å2 Details: Images were recorded in super-resolution mode with 40 frames per image |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2.7 mm / Nominal defocus max: 3.0 µm / Nominal defocus min: 1.8 µm / Nominal magnification: 81000 |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
+Image processing
-Atomic model buiding 1
| Refinement | Space: REAL / Protocol: FLEXIBLE FIT / Target criteria: Correlation coefficient |
|---|
