Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-13185 | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | Helical structure of the toxin MakA from Vibrio cholera | |||||||||||||||
 Map data Map data | ||||||||||||||||
 Sample Sample |
| |||||||||||||||
 Keywords Keywords | Pore-forming toxin / Vibrio cholerae / TOXIN | |||||||||||||||
| Function / homology | Hemolysin BL-binding component / Bacillus haemolytic enterotoxin (HBL) / : / membrane / Non-hemolytic enterotoxin lytic component L1 Function and homology information Function and homology information | |||||||||||||||
| Biological species |  Vibrio cholerae O1 biovar El Tor str. N16961 (bacteria) / Vibrio cholerae O1 biovar El Tor str. N16961 Vibrio cholerae O1 biovar El Tor str. N16961 (bacteria) / Vibrio cholerae O1 biovar El Tor str. N16961 | |||||||||||||||
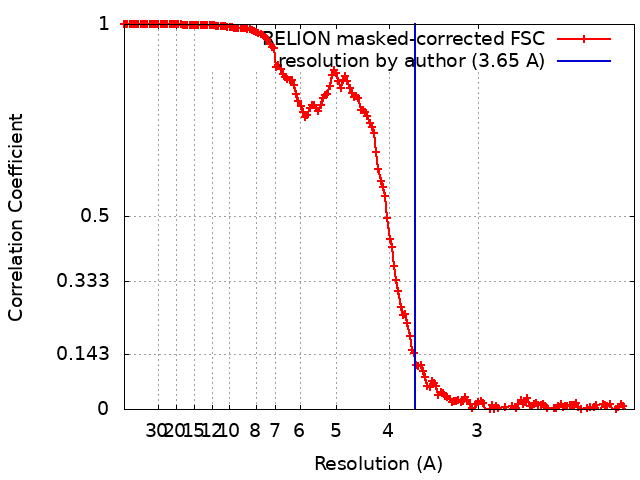
| Method | helical reconstruction / cryo EM / Resolution: 3.65 Å | |||||||||||||||
 Authors Authors | Berg A / Nadeem A | |||||||||||||||
| Funding support |  Sweden, European Union, 4 items Sweden, European Union, 4 items
| |||||||||||||||
 Citation Citation | Journal: Elife / Year: 2022 Title: Protein-lipid interaction at low pH induces oligomerization of the MakA cytotoxin from . Authors: Aftab Nadeem / Alexandra Berg / Hudson Pace / Athar Alam / Eric Toh / Jörgen Ådén / Nikola Zlatkov / Si Lhyam Myint / Karina Persson / Gerhard Gröbner / Anders Sjöstedt / Marta Bally / ...Authors: Aftab Nadeem / Alexandra Berg / Hudson Pace / Athar Alam / Eric Toh / Jörgen Ådén / Nikola Zlatkov / Si Lhyam Myint / Karina Persson / Gerhard Gröbner / Anders Sjöstedt / Marta Bally / Jonas Barandun / Bernt Eric Uhlin / Sun Nyunt Wai / Abstract: The α-pore-forming toxins (α-PFTs) from pathogenic bacteria damage host cell membranes by pore formation. We demonstrate a remarkable, hitherto unknown mechanism by an α-PFT protein from . As part ...The α-pore-forming toxins (α-PFTs) from pathogenic bacteria damage host cell membranes by pore formation. We demonstrate a remarkable, hitherto unknown mechanism by an α-PFT protein from . As part of the MakA/B/E tripartite toxin, MakA is involved in membrane pore formation similar to other α-PFTs. In contrast, MakA in isolation induces tube-like structures in acidic endosomal compartments of epithelial cells in vitro. The present study unravels the dynamics of tubular growth, which occurs in a pH-, lipid-, and concentration-dependent manner. Within acidified organelle lumens or when incubated with cells in acidic media, MakA forms oligomers and remodels membranes into high-curvature tubes leading to loss of membrane integrity. A 3.7 Å cryo-electron microscopy structure of MakA filaments reveals a unique protein-lipid superstructure. MakA forms a pinecone-like spiral with a central cavity and a thin annular lipid bilayer embedded between the MakA transmembrane helices in its active α-PFT conformation. Our study provides insights into a novel tubulation mechanism of an α-PFT protein and a new mode of action by a secreted bacterial toxin. | |||||||||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |

- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_13185.map.gz | 342.8 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-13185-v30.xmlemd-13185.xml | 19.4 KB 19.4 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_13185_fsc.xml | 16.3 KB | Display | FSC data file |
| Images |  emd_13185.png emd_13185.png | 213 KB | ||
| Filedesc metadata | emd-13185.cif.gz | 6 KB | ||
| Others | emd_13185_additional_1.map.gzemd_13185_half_map_1.map.gzemd_13185_half_map_2.map.gz | 5.9 MB 295.8 MB 295.8 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-13185ftp://ftp.pdbj.org/pub/emdb/structures/EMD-13185 http://ftp.pdbj.org/pub/emdb/structures/EMD-13185ftp://ftp.pdbj.org/pub/emdb/structures/EMD-13185 | HTTPS FTP |
-Validation report
| Summary document | emd_13185_validation.pdf.gz | 1.2 MB | Display | EMDB validaton report |
|---|---|---|---|---|
| Full document | emd_13185_full_validation.pdf.gz | 1.2 MB | Display | |
| Data in XML | emd_13185_validation.xml.gz | 23.8 KB | Display | |
| Data in CIF | emd_13185_validation.cif.gz | 31.8 KB | Display | |
| Arichive directory | https://ftp.pdbj.org/pub/emdb/validation_reports/EMD-13185ftp://ftp.pdbj.org/pub/emdb/validation_reports/EMD-13185 | HTTPS FTP |
-Related structure data
| Related structure data |  7p3rMC M: atomic model generated by this map C: citing same article ( |
|---|---|
| Similar structure data | |
| EM raw data | EMPIAR-10869 (Title: Helical structure of the toxin MakA from Vibrio cholera Data size: 195.8 Data #1: Unaligned multi-frame micrographs of the oligomerized Vibrio cholerae toxin MakA [micrographs - multiframe]) |






-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_13185.map.gz / Format: CCP4 / Size: 371.3 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
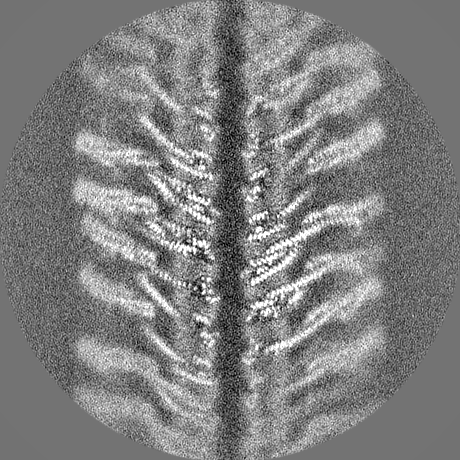
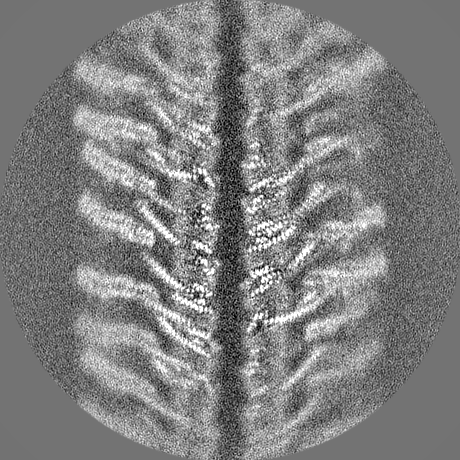
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.042 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
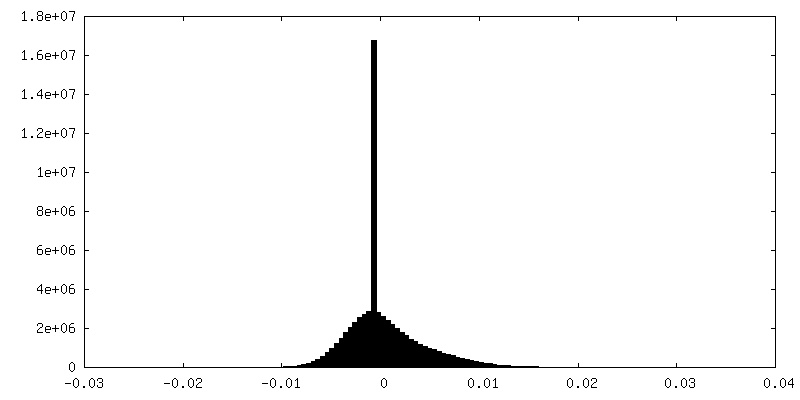
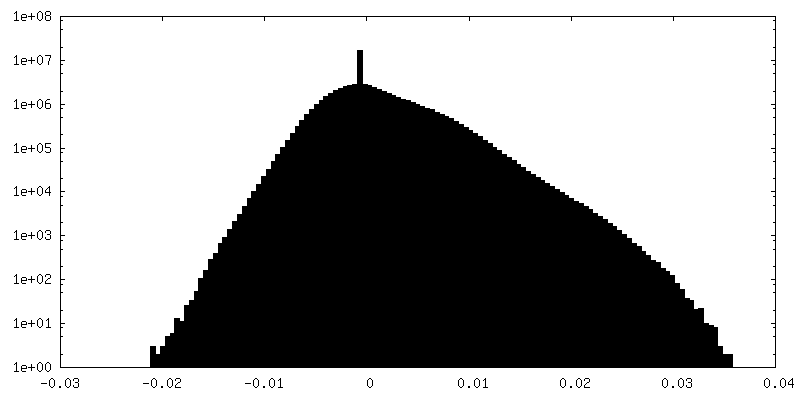
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















-Supplemental data
-Additional map: additional map 1
| File | emd_13185_additional_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | additional map 1 | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |






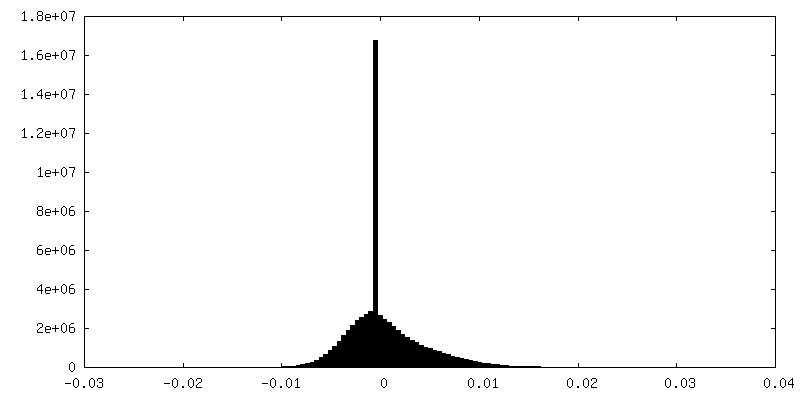
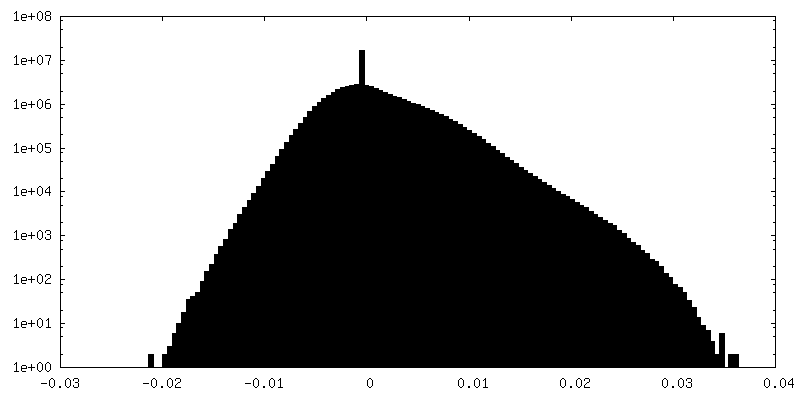
-Half map: hafl map 2
| File | emd_13185_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | hafl map 2 | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |

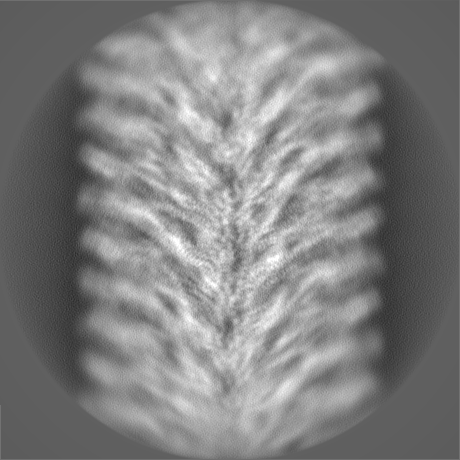
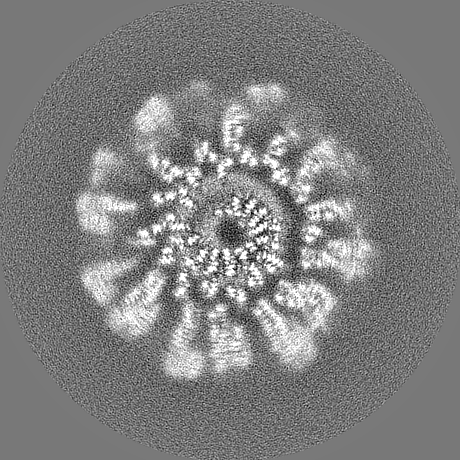
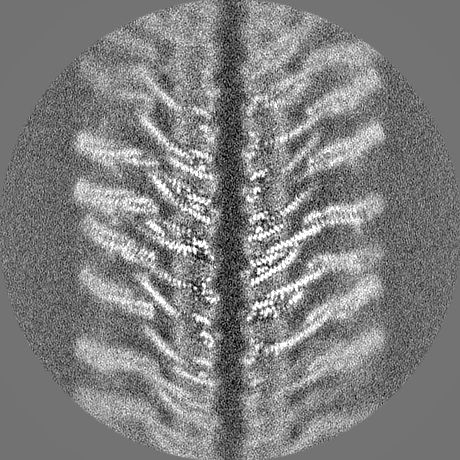
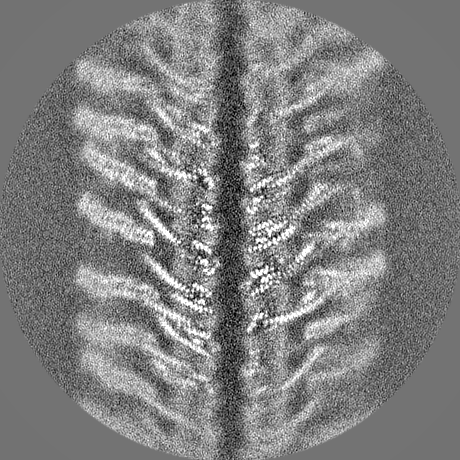
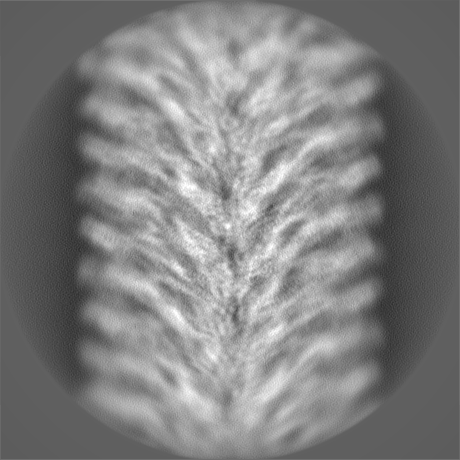
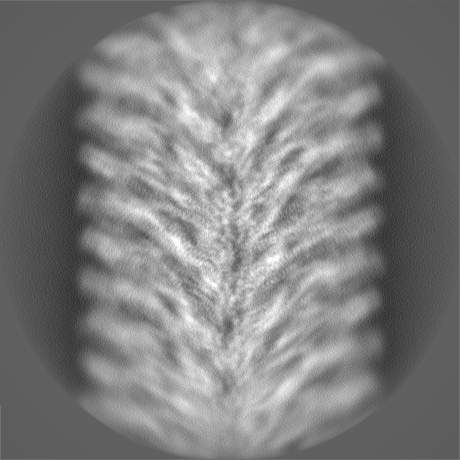
-Half map: hafl map 1
| File | emd_13185_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | hafl map 1 | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |


- Sample components
Sample components
-Entire : Vibrio cholera MakA filament
| Entire | Name: Vibrio cholera MakA filament |
|---|---|
| Components |
|
-Supramolecule #1: Vibrio cholera MakA filament
| Supramolecule | Name: Vibrio cholera MakA filament / type: complex / ID: 1 / Parent: 0 / Macromolecule list: all |
|---|---|
| Source (natural) | Organism: Vibrio cholerae O1 biovar El Tor str. N16961 (bacteria) |
-Macromolecule #1: MakA tetramer
| Macromolecule | Name: MakA tetramer / type: protein_or_peptide / ID: 1 / Number of copies: 4 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: Vibrio cholerae O1 biovar El Tor str. N16961 |
| Molecular weight | Theoretical: 39.028582 KDa |
| Recombinant expression | Organism: |
| Sequence | String: MSQQVTQLNP TQQTTQSAFL ATTVITAQCH AILNTQFTPP TVKPDWFDDL SKKLDSAKLV AKQWIDDLGP QVSASIPSSV INFDATFQA SIDAIHELYK ADPTASGKDN TTVQQASQIM TALSSQVSGI EATVKGMNKE LSDWGVKMQA AHDDLVNGAT N IQKTIIDL ...String: MSQQVTQLNP TQQTTQSAFL ATTVITAQCH AILNTQFTPP TVKPDWFDDL SKKLDSAKLV AKQWIDDLGP QVSASIPSSV INFDATFQA SIDAIHELYK ADPTASGKDN TTVQQASQIM TALSSQVSGI EATVKGMNKE LSDWGVKMQA AHDDLVNGAT N IQKTIIDL QTDIESMNNA IDNNRAAIEK LNKDLVYAQV AVGVGIFMLV AGVALTVATA GTAAAVSGGI AAVGAASIIA GG VTWGVLQ NQIDDDYDSI AQEQKQKAED QQQIIALQGL SNASSAVVSA IETSTSVLSD FETTWTVFGN ELDDVVTKLN NGA SMQSII MEKVMSDAAK NEWDDAVELA KQLASAKIAI ETKELAPAVK QAA UniProtKB: Non-hemolytic enterotoxin lytic component L1 |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | helical reconstruction |
| Aggregation state | helical array |
-Sample preparation
| Buffer | pH: 6.5 / Component - Concentration: 120.0 mM / Component - Name: Sodium Citrate |
|---|---|
| Grid | Model: Quantifoil R2/1 / Material: COPPER / Mesh: 200 / Pretreatment - Type: GLOW DISCHARGE |
| Vitrification | Cryogen name: ETHANE / Instrument: FEI VITROBOT MARK I |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Image recording | Film or detector model: GATAN K2 QUANTUM (4k x 4k) / Detector mode: COUNTING / Number grids imaged: 1 / Number real images: 2476 / Average electron dose: 43.0 e/Å2 |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2.7 mm / Nominal defocus max: 2.5 µm / Nominal defocus min: 0.75 µm |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER / Cooling holder cryogen: NITROGEN |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |