 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
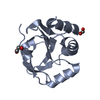
Yorodumi- PDB-1lu4: 1.1 ANGSTROM RESOLUTION CRYSTAL STRUCTURE OF A SECRETED MYCOBACTE... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1lu4 | ||||||
|---|---|---|---|---|---|---|---|
| Title | 1.1 ANGSTROM RESOLUTION CRYSTAL STRUCTURE OF A SECRETED MYCOBACTERIUM TUBERCULOSIS DISULFIDE OXIDOREDUCTASE HOMOLOGOUS TO E. COLI DSBE: IMPLICATIONS FOR FUNCTIONS | ||||||
 Components Components | SOLUBLE SECRETED ANTIGEN MPT53 | ||||||
 Keywords Keywords | OXIDOREDUCTASE / THIOREDOXIN-LIKE FOLD / Structural Genomics / PSI / Protein Structure Initiative / TB Structural Genomics Consortium / TBSGC | ||||||
| Function / homology |  Function and homology information Function and homology informationantioxidant activity / cell redox homeostasis / oxidoreductase activity / extracellular region Similarity search - Function | ||||||
| Biological species |   Mycobacterium tuberculosis (bacteria) Mycobacterium tuberculosis (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / AB INITIO / Resolution: 1.12 Å X-RAY DIFFRACTION / SYNCHROTRON / AB INITIO / Resolution: 1.12 Å | ||||||
 Authors Authors | Goulding, C.W. / Apostol, M.I. / Gleiter, S. / Parseghian, A. / Bardwell, J. / Gennaro, M. / Eisenberg, D. / TB Structural Genomics Consortium (TBSGC) | ||||||
 Citation Citation | Journal: J.Biol.Chem. / Year: 2004 Title: Gram-positive DsbE Proteins Function Differently from Gram-negative DsbE Homologs: A STRUCTURE TO FUNCTION ANALYSIS OF DsbE FROM MYCOBACTERIUM TUBERCULOSIS. Authors: Goulding, C.W. / Apostol, M.I. / Gleiter, S. / Parseghian, A. / Bardwell, J. / Gennaro, M. / Eisenberg, D. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1lu4.cif.gz | 47.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1lu4.ent.gz | 33.2 KB | Display | PDB format |
| PDBx/mmJSON format | 1lu4.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 1lu4_validation.pdf.gz | 401.1 KB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 1lu4_full_validation.pdf.gz | 407.9 KB | Display | |
| Data in XML | 1lu4_validation.xml.gz | 6.1 KB | Display | |
| Data in CIF | 1lu4_validation.cif.gz | 9.6 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/lu/1lu4ftp://data.pdbj.org/pub/pdb/validation_reports/lu/1lu4 | HTTPS FTP |
-Related structure data
| Similar structure data | |
|---|---|
| Other databases |









-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| |||||||||||||||
| Unit cell |
| |||||||||||||||
| Components on special symmetry positions |
| |||||||||||||||
| Details | monomer in asymmetric unit |
-Components
| #1: Protein | Mass: 14624.241 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Mycobacterium tuberculosis (bacteria) / Gene: Rv2878c / Plasmid: pQE30 / Production host: |
|---|---|
| #2: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 321 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 321 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.18 Å3/Da / Density % sol: 43.6 % | ||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 300 K / Method: vapor diffusion, hanging drop / pH: 7.4 Details: 2.2M ammonium sulfate, 5% isopropanol, 20% glycerol, pH 7.4, VAPOR DIFFUSION, HANGING DROP, temperature 300K | ||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Method: vapor diffusion | ||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 200 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: NSLS  / Beamline: X8C / Wavelength: 0.9792 Å / Beamline: X8C / Wavelength: 0.9792 Å |
| Detector | Type: ADSC QUANTUM 4 / Detector: CCD / Details: mirrors |
| Radiation | Monochromator: Si III channel / Protocol: MAD / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9792 Å / Relative weight: 1 |
| Reflection | Resolution: 1.12→19.99 Å / Num. all: 57093 / Num. obs: 57039 / % possible obs: 99.99 % / Observed criterion σ(F): 1 / Observed criterion σ(I): 1 / Redundancy: 8.3 % / Rmerge(I) obs: 0.074 / Net I/σ(I): 6.8 |
| Reflection shell | Resolution: 1.12→1.19 Å / Rmerge(I) obs: 0.227 / Mean I/σ(I) obs: 11.4 / Num. unique all: 5671 / % possible all: 100 |
| Reflection | *PLUS Highest resolution: 1.5 Å / Num. obs: 45334 / % possible obs: 99.9 % / Num. measured all: 285221 / Rmerge(I) obs: 0.088 |
| Reflection shell | *PLUS % possible obs: 100 % / Rmerge(I) obs: 0.457 / Mean I/σ(I) obs: 3.2 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: AB INITIO / Resolution: 1.12→10 Å / Num. parameters: 12486 / Num. restraintsaints: 15009 / Cross valid method: FREE R / σ(F): 0 / Stereochemistry target values: ENGH AND HUBER Details: ANISOTROPIC REFINEMENT REDUCED FREE R (NO CUTOFF) BY ?
| |||||||||||||||||||||||||||||||||
| Refine analyze | Num. disordered residues: 7 / Occupancy sum hydrogen: 0 / Occupancy sum non hydrogen: 1328 | |||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.12→10 Å
| |||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||
| Software | *PLUS Name: SHELXL / Version: 97 / Classification: refinement | |||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 1.1 Å / Lowest resolution: 20 Å / % reflection Rfree: 5 % / Rfactor Rfree: 0.203 / Rfactor Rwork: 0.143 | |||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | |||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | |||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
