 ムービー
ムービー コントローラー
コントローラー


+ データを開く
データを開く

- 基本情報
基本情報
| 登録情報 | データベース: PDB / ID: 1abb | ||||||
|---|---|---|---|---|---|---|---|
| タイトル | CONTROL OF PHOSPHORYLASE B CONFORMATION BY A MODIFIED COFACTOR: CRYSTALLOGRAPHIC STUDIES ON R-STATE GLYCOGEN PHOSPHORYLASE RECONSTITUTED WITH PYRIDOXAL 5'-DIPHOSPHATE | ||||||
 要素 要素 | GLYCOGEN PHOSPHORYLASE B | ||||||
 キーワード キーワード | GLYCOGEN PHOSPHORYLASE | ||||||
| 機能・相同性 |  機能・相同性情報 機能・相同性情報glycogen phosphorylase / glycogen phosphorylase activity / linear malto-oligosaccharide phosphorylase activity / SHG alpha-glucan phosphorylase activity / glycogen catabolic process / skeletal muscle myofibril / pyridoxal phosphate binding / nucleotide binding 類似検索 - 分子機能 | ||||||
| 生物種 |  | ||||||
| 手法 |  X線回折 / 解像度: 2.8 Å X線回折 / 解像度: 2.8 Å | ||||||
 データ登録者 データ登録者 | Leonidas, D.D. / Oikonomakos, N.G. / Papageorgiou, A.C. / Acharya, K.R. / Barford, D. / Johnson, L.N. | ||||||
 引用 引用 | ジャーナル: Protein Sci. / 年: 1992 タイトル: Control of phosphorylase b conformation by a modified cofactor: crystallographic studies on R-state glycogen phosphorylase reconstituted with pyridoxal 5'-diphosphate. 著者: Leonidas, D.D. / Oikonomakos, N.G. / Papageorgiou, A.C. / Acharya, K.R. / Barford, D. / Johnson, L.N. #1: ジャーナル: Nature / 年: 1989タイトル: The Allosteric Transition of Glycogen Phosphorylase 著者: Barford, D. / Johnson, L.N. | ||||||
| 履歴 |
|
- 構造の表示
構造の表示
| 構造ビューア | 分子: MolmilJmol/JSmol |
|---|
- ダウンロードとリンク
ダウンロードとリンク
-ダウンロード
| PDBx/mmCIF形式 | 1abb.cif.gz | 667.7 KB | 表示 | PDBx/mmCIF形式 |
|---|---|---|---|---|
| PDB形式 | pdb1abb.ent.gz | 539.2 KB | 表示 | PDB形式 |
| PDBx/mmJSON形式 | 1abb.json.gz | ツリー表示 | PDBx/mmJSON形式 | |
| その他 |  その他のダウンロード その他のダウンロード |
-検証レポート
| 文書・要旨 | 1abb_validation.pdf.gz | 770.3 KB | 表示 | wwPDB検証レポート |
|---|---|---|---|---|
| 文書・詳細版 | 1abb_full_validation.pdf.gz | 1017.9 KB | 表示 | |
| XML形式データ | 1abb_validation.xml.gz | 94.5 KB | 表示 | |
| CIF形式データ | 1abb_validation.cif.gz | 131.8 KB | 表示 | |
| アーカイブディレクトリ | https://data.pdbj.org/pub/pdb/validation_reports/ab/1abbftp://data.pdbj.org/pub/pdb/validation_reports/ab/1abb | HTTPS FTP |
-関連構造データ










-リンク
 PDBj
PDBj
- 集合体
集合体
| 登録構造単位 | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| 単位格子 |
| ||||||||
| Atom site foot note | 1: TYR 1 280 - PRO 1 281 OMEGA ANGLE = 275.327 PEPTIDE BOND DEVIATES SIGNIFICANTLY FROM TRANS CONFORMATION 2: ASP 1 320 - PRO 1 321 OMEGA ANGLE = 231.150 PEPTIDE BOND DEVIATES SIGNIFICANTLY FROM TRANS CONFORMATION 3: ALA 1 836 - PRO 1 837 OMEGA ANGLE = 329.984 PEPTIDE BOND DEVIATES SIGNIFICANTLY FROM TRANS CONFORMATION 4: TRP 2 67 - ILE 2 68 OMEGA ANGLE = 138.728 PEPTIDE BOND DEVIATES SIGNIFICANTLY FROM TRANS CONFORMATION 5: TYR 2 280 - PRO 2 281 OMEGA ANGLE = 270.308 PEPTIDE BOND DEVIATES SIGNIFICANTLY FROM TRANS CONFORMATION 6: ASP 2 320 - PRO 2 321 OMEGA ANGLE = 228.278 PEPTIDE BOND DEVIATES SIGNIFICANTLY FROM TRANS CONFORMATION 7: CIS PROLINE - PRO 2 837 8: TYR 3 280 - PRO 3 281 OMEGA ANGLE = 275.626 PEPTIDE BOND DEVIATES SIGNIFICANTLY FROM TRANS CONFORMATION 9: ASP 3 320 - PRO 3 321 OMEGA ANGLE = 249.790 PEPTIDE BOND DEVIATES SIGNIFICANTLY FROM TRANS CONFORMATION 10: ALA 3 836 - PRO 3 837 OMEGA ANGLE = 314.024 PEPTIDE BOND DEVIATES SIGNIFICANTLY FROM TRANS CONFORMATION 11: TYR 4 280 - PRO 4 281 OMEGA ANGLE = 267.181 PEPTIDE BOND DEVIATES SIGNIFICANTLY FROM TRANS CONFORMATION 12: ASP 4 320 - PRO 4 321 OMEGA ANGLE = 238.142 PEPTIDE BOND DEVIATES SIGNIFICANTLY FROM TRANS CONFORMATION 13: ALA 4 836 - PRO 4 837 OMEGA ANGLE = 317.052 PEPTIDE BOND DEVIATES SIGNIFICANTLY FROM TRANS CONFORMATION |
-要素
| #1: タンパク質 | 分子量: 95664.398 Da / 分子数: 4 / 由来タイプ: 組換発現 / 由来: (組換発現) #2: 化合物 | ChemComp-SO4 /   分子量: 96.063 Da / 分子数: 4 / 由来タイプ: 合成 / 式: SO4 分子量: 96.063 Da / 分子数: 4 / 由来タイプ: 合成 / 式: SO4#3: 化合物 | ChemComp-PDP / 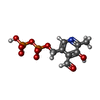  分子量: 327.122 Da / 分子数: 4 / 由来タイプ: 合成 / 式: C8H11NO9P2 分子量: 327.122 Da / 分子数: 4 / 由来タイプ: 合成 / 式: C8H11NO9P2#4: 化合物 | ChemComp-IMP / 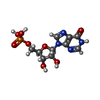  分子量: 348.206 Da / 分子数: 4 / 由来タイプ: 合成 / 式: C10H13N4O8P 分子量: 348.206 Da / 分子数: 4 / 由来タイプ: 合成 / 式: C10H13N4O8P配列の詳細 | RESIDUE 380 WAS BUILT AS AN ISOLEUCINE AS THIS SIDE CHAIN APPEARED TO FIT THE ELECTRON DENSITY FOR ...RESIDUE 380 WAS BUILT AS AN ISOLEUCINE | |
|---|
-実験情報
-実験
| 実験 | 手法: X線回折 |
|---|
- 試料調製
試料調製
| 結晶 | マシュー密度: 2.43 Å3/Da / 溶媒含有率: 49.41 % | ||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 結晶化 | *PLUS 温度: 16 ℃ / pH: 7.5 / 手法: microdialysis | ||||||||||||||||||||||||||||||||||||||||||||||||
| 溶液の組成 | *PLUS
|
-データ収集
| 放射 | 散乱光タイプ: x-ray |
|---|---|
| 放射波長 | 相対比: 1 |
| 反射 | *PLUS 最高解像度: 2.8 Å / Num. obs: 78049 / % possible obs: 98 % / Num. measured all: 107286 / Rmerge(I) obs: 0.108 |
- 解析
解析
| ソフトウェア |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 精密化 | 解像度: 2.8→8 Å / σ(F): 1 /
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 精密化ステップ | サイクル: LAST / 解像度: 2.8→8 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 拘束条件 |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 精密化 | *PLUS Num. reflection obs: 73340 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 溶媒の処理 | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 原子変位パラメータ | *PLUS |
