 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1abb: CONTROL OF PHOSPHORYLASE B CONFORMATION BY A MODIFIED COFACTOR: C... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1abb | ||||||
|---|---|---|---|---|---|---|---|
| Title | CONTROL OF PHOSPHORYLASE B CONFORMATION BY A MODIFIED COFACTOR: CRYSTALLOGRAPHIC STUDIES ON R-STATE GLYCOGEN PHOSPHORYLASE RECONSTITUTED WITH PYRIDOXAL 5'-DIPHOSPHATE | ||||||
 Components Components | GLYCOGEN PHOSPHORYLASE B | ||||||
 Keywords Keywords | GLYCOGEN PHOSPHORYLASE | ||||||
| Function / homology |  Function and homology information Function and homology informationglycogen phosphorylase / glycogen phosphorylase activity / glycogen catabolic process / skeletal muscle myofibril / pyridoxal phosphate binding / nucleotide binding Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / Resolution: 2.8 Å X-RAY DIFFRACTION / Resolution: 2.8 Å | ||||||
 Authors Authors | Leonidas, D.D. / Oikonomakos, N.G. / Papageorgiou, A.C. / Acharya, K.R. / Barford, D. / Johnson, L.N. | ||||||
 Citation Citation | Journal: Protein Sci. / Year: 1992 Title: Control of phosphorylase b conformation by a modified cofactor: crystallographic studies on R-state glycogen phosphorylase reconstituted with pyridoxal 5'-diphosphate. Authors: Leonidas, D.D. / Oikonomakos, N.G. / Papageorgiou, A.C. / Acharya, K.R. / Barford, D. / Johnson, L.N. #1: Journal: Nature / Year: 1989Title: The Allosteric Transition of Glycogen Phosphorylase Authors: Barford, D. / Johnson, L.N. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1abb.cif.gz | 668.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1abb.ent.gz | 539.2 KB | Display | PDB format |
| PDBx/mmJSON format | 1abb.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ab/1abbftp://data.pdbj.org/pub/pdb/validation_reports/ab/1abb | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|










-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Atom site foot note | 1: TYR 1 280 - PRO 1 281 OMEGA ANGLE = 275.327 PEPTIDE BOND DEVIATES SIGNIFICANTLY FROM TRANS CONFORMATION 2: ASP 1 320 - PRO 1 321 OMEGA ANGLE = 231.150 PEPTIDE BOND DEVIATES SIGNIFICANTLY FROM TRANS CONFORMATION 3: ALA 1 836 - PRO 1 837 OMEGA ANGLE = 329.984 PEPTIDE BOND DEVIATES SIGNIFICANTLY FROM TRANS CONFORMATION 4: TRP 2 67 - ILE 2 68 OMEGA ANGLE = 138.728 PEPTIDE BOND DEVIATES SIGNIFICANTLY FROM TRANS CONFORMATION 5: TYR 2 280 - PRO 2 281 OMEGA ANGLE = 270.308 PEPTIDE BOND DEVIATES SIGNIFICANTLY FROM TRANS CONFORMATION 6: ASP 2 320 - PRO 2 321 OMEGA ANGLE = 228.278 PEPTIDE BOND DEVIATES SIGNIFICANTLY FROM TRANS CONFORMATION 7: CIS PROLINE - PRO 2 837 8: TYR 3 280 - PRO 3 281 OMEGA ANGLE = 275.626 PEPTIDE BOND DEVIATES SIGNIFICANTLY FROM TRANS CONFORMATION 9: ASP 3 320 - PRO 3 321 OMEGA ANGLE = 249.790 PEPTIDE BOND DEVIATES SIGNIFICANTLY FROM TRANS CONFORMATION 10: ALA 3 836 - PRO 3 837 OMEGA ANGLE = 314.024 PEPTIDE BOND DEVIATES SIGNIFICANTLY FROM TRANS CONFORMATION 11: TYR 4 280 - PRO 4 281 OMEGA ANGLE = 267.181 PEPTIDE BOND DEVIATES SIGNIFICANTLY FROM TRANS CONFORMATION 12: ASP 4 320 - PRO 4 321 OMEGA ANGLE = 238.142 PEPTIDE BOND DEVIATES SIGNIFICANTLY FROM TRANS CONFORMATION 13: ALA 4 836 - PRO 4 837 OMEGA ANGLE = 317.052 PEPTIDE BOND DEVIATES SIGNIFICANTLY FROM TRANS CONFORMATION |
-Components
| #1: Protein | Mass: 95664.398 Da / Num. of mol.: 4 Source method: isolated from a genetically manipulated source Source: (gene. exp.) #2: Chemical | ChemComp-SO4 /   Mass: 96.063 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: SO4#3: Chemical | ChemComp-PDP / 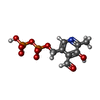  Mass: 327.122 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C8H11NO9P2 Mass: 327.122 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C8H11NO9P2#4: Chemical | ChemComp-IMP / 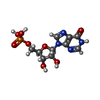  Mass: 348.206 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C10H13N4O8P Mass: 348.206 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C10H13N4O8PHas protein modification | Y | Sequence details | RESIDUE 380 WAS BUILT AS AN ISOLEUCINE AS THIS SIDE CHAIN APPEARED TO FIT THE ELECTRON DENSITY FOR ...RESIDUE 380 WAS BUILT AS AN ISOLEUCINE | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.43 Å3/Da / Density % sol: 49.41 % | ||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | *PLUS Temperature: 16 ℃ / pH: 7.5 / Method: microdialysis | ||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Radiation | Scattering type: x-ray |
|---|---|
| Radiation wavelength | Relative weight: 1 |
| Reflection | *PLUS Highest resolution: 2.8 Å / Num. obs: 78049 / % possible obs: 98 % / Num. measured all: 107286 / Rmerge(I) obs: 0.108 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 2.8→8 Å / σ(F): 1 /
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.8→8 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Num. reflection obs: 73340 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS |
