 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 5os7 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | The crystal structure of CK2alpha in complex with compound 4 | |||||||||
 Components Components | Casein kinase II subunit alpha | |||||||||
 Keywords Keywords | TRANSFERASE / CK2alpha / CK2a / fragment based drug discovery / high concentration screening / selective ATP competitive inhibitors / surface entrophy reduction | |||||||||
| Function / homology |  Function and homology information Function and homology informationPhosphorylation and nuclear translocation of BMAL1 (ARNTL) and CLOCK / positive regulation of aggrephagy / regulation of chromosome separation / WNT mediated activation of DVL / Condensation of Prometaphase Chromosomes / protein kinase CK2 complex / symbiont-mediated disruption of host cell PML body / Phosphorylation and nuclear translocation of the CRY:PER:kinase complex / Regulation of CDH1 posttranslational processing and trafficking to plasma membrane / Receptor Mediated Mitophagy ...Phosphorylation and nuclear translocation of BMAL1 (ARNTL) and CLOCK / positive regulation of aggrephagy / regulation of chromosome separation / WNT mediated activation of DVL / Condensation of Prometaphase Chromosomes / protein kinase CK2 complex / symbiont-mediated disruption of host cell PML body / Phosphorylation and nuclear translocation of the CRY:PER:kinase complex / Regulation of CDH1 posttranslational processing and trafficking to plasma membrane / Receptor Mediated Mitophagy / Synthesis of PC / Sin3-type complex / negative regulation of signal transduction by p53 class mediator / RUNX1 interacts with co-factors whose precise effect on RUNX1 targets is not known / Maturation of hRSV A proteins / negative regulation of apoptotic signaling pathway / negative regulation of double-strand break repair via homologous recombination / positive regulation of Wnt signaling pathway / negative regulation of proteasomal ubiquitin-dependent protein catabolic process / Signal transduction by L1 / Hsp90 protein binding / PML body / Wnt signaling pathway / Regulation of PTEN stability and activity / kinase activity / positive regulation of protein catabolic process / KEAP1-NFE2L2 pathway / rhythmic process / double-strand break repair / Cooperation of PDCL (PhLP1) and TRiC/CCT in G-protein beta folding / positive regulation of cell growth / protein folding / Regulation of TP53 Activity through Phosphorylation / non-specific serine/threonine protein kinase / regulation of cell cycle / negative regulation of translation / protein stabilization / protein serine kinase activity / protein serine/threonine kinase activity / apoptotic process / positive regulation of cell population proliferation / DNA damage response / signal transduction / nucleoplasm / ATP binding / identical protein binding / nucleus / plasma membrane / cytosol Similarity search - Function | |||||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | |||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / Resolution: 1.66 Å X-RAY DIFFRACTION / SYNCHROTRON / Resolution: 1.66 Å | |||||||||
 Authors Authors | Brear, P. / De Fusco, C. / Iegre, J. / Yoshida, M. / Mitchell, S. / Rossmann, M. / Carro, L. / Sore, H. / Hyvonen, M. / Spring, D. | |||||||||
| Funding support |  United Kingdom, 2items United Kingdom, 2items
| |||||||||
 Citation Citation | Journal: Chem Sci / Year: 2018 Title: Second-generation CK2 alpha inhibitors targeting the alpha D pocket. Authors: Iegre, J. / Brear, P. / De Fusco, C. / Yoshida, M. / Mitchell, S.L. / Rossmann, M. / Carro, L. / Sore, H.F. / Hyvonen, M. / Spring, D.R. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 5os7.cif.gz | 165 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb5os7.ent.gz | 127.9 KB | Display | PDB format |
| PDBx/mmJSON format | 5os7.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/os/5os7ftp://data.pdbj.org/pub/pdb/validation_reports/os/5os7 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  5oquC  5orhC  5orjC  5orkC  5os8C  5oslC  5ospC  5osrC  5osuC  5oszC  5ot5C  5ot6C  5otdC  5othC  5otiC  5otlC  5otoC  5otpC  5otqC  5otrC  5otsC  5otyC  5otzC  5oueC  5oulC  5oumC  5ouuC  5oyfC  6ehkC  6ehuC  6eiiC  5cvhS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |




















-Links
 PDBj
PDBj




- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 41467.793 Da / Num. of mol.: 2 Fragment: residues 2-329 and N-terminal extension GSMDIEFDDDADDDGSGSGSGSGS Mutation: R21S Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: CSNK2A1, CK2A1 / Production host:  References: UniProt: P68400, non-specific serine/threonine protein kinase #2: Chemical | ChemComp-ACT /   Mass: 59.044 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C2H3O2 Mass: 59.044 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C2H3O2#3: Chemical | 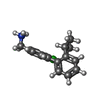  Mass: 260.782 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C16H19ClN / Feature type: SUBJECT OF INVESTIGATION Mass: 260.782 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C16H19ClN / Feature type: SUBJECT OF INVESTIGATION#4: Chemical |   Mass: 507.181 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H16N5O13P3 / Comment: ATP, energy-carrying molecule*YM Mass: 507.181 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H16N5O13P3 / Comment: ATP, energy-carrying molecule*YM#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 405 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 405 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.24 Å3/Da / Density % sol: 45.05 % / Mosaicity: 0 ° |
|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, hanging drop / pH: 6.5 Details: 112.5mM Mes pH 6.5, 35% glycerol ethoxylate, 180 mM ammonium acetate |
-Data collection
| Diffraction | Mean temperature: 100 K | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: Diamond / Beamline: I24 / Wavelength: 0.96862 Å | |||||||||||||||||||||
| Detector | Type: DECTRIS PILATUS 6M / Detector: PIXEL / Date: Nov 25, 2015 | |||||||||||||||||||||
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | |||||||||||||||||||||
| Radiation wavelength | Wavelength: 0.96862 Å / Relative weight: 1 | |||||||||||||||||||||
| Reflection | Resolution: 1.66→66.65 Å / Num. obs: 88560 / % possible obs: 100 % / Redundancy: 7.8 % / Biso Wilson estimate: 18.05 Å2 / CC1/2: 0.994 / Rmerge(I) obs: 0.13 / Rpim(I) all: 0.05 / Rrim(I) all: 0.139 / Net I/σ(I): 8.1 / Num. measured all: 687678 / Scaling rejects: 2317 | |||||||||||||||||||||
| Reflection shell | Diffraction-ID: 1
|
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Starting model: 5CVH Resolution: 1.66→55.54 Å / Cor.coef. Fo:Fc: 0.9156 / Cor.coef. Fo:Fc free: 0.9018 / SU R Cruickshank DPI: 0.113 / Cross valid method: THROUGHOUT / σ(F): 0 / SU R Blow DPI: 0.115 / SU Rfree Blow DPI: 0.108 / SU Rfree Cruickshank DPI: 0.108
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 122.04 Å2 / Biso mean: 25.53 Å2 / Biso min: 3.84 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.264 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 1.66→55.54 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.66→1.7 Å / Rfactor Rfree error: 0 / Total num. of bins used: 20
|
