 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-5oip: InhA (T2A mutant) complexed with 1-(Pyridin-3-ylmethyl)-3-(1-(pyr... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 5oip | ||||||
|---|---|---|---|---|---|---|---|
| Title | InhA (T2A mutant) complexed with 1-(Pyridin-3-ylmethyl)-3-(1-(pyrimidin-2-yl)piperidin-4-yl)urea | ||||||
 Components Components | Enoyl-[acyl-carrier-protein] reductase [NADH] | ||||||
 Keywords Keywords | OXIDOREDUCTASE / Inhibitor / complex / fragment based drug discovery / tuberculosis | ||||||
| Function / homology |  Function and homology information Function and homology informationtrans-2-enoyl-CoA reductase (NADH) activity / mycolic acid biosynthetic process / enoyl-[acyl-carrier-protein] reductase (NADH) / fatty acid elongation / enoyl-[acyl-carrier-protein] reductase (NADH) activity / NAD+ binding / peptidoglycan-based cell wall / fatty acid binding / response to antibiotic / plasma membrane Similarity search - Function | ||||||
| Biological species |   Mycobacterium tuberculosis (bacteria) Mycobacterium tuberculosis (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / Resolution: 1.71 Å X-RAY DIFFRACTION / SYNCHROTRON / Resolution: 1.71 Å | ||||||
 Authors Authors | Convery, M.A. | ||||||
 Citation Citation | Journal: ChemMedChem / Year: 2018 Title: Screening of a Novel Fragment Library with Functional Complexity against Mycobacterium tuberculosis InhA. Authors: Prati, F. / Zuccotto, F. / Fletcher, D. / Convery, M.A. / Fernandez-Menendez, R. / Bates, R. / Encinas, L. / Zeng, J. / Chung, C.W. / De Dios Anton, P. / Mendoza-Losana, A. / Mackenzie, C. / ...Authors: Prati, F. / Zuccotto, F. / Fletcher, D. / Convery, M.A. / Fernandez-Menendez, R. / Bates, R. / Encinas, L. / Zeng, J. / Chung, C.W. / De Dios Anton, P. / Mendoza-Losana, A. / Mackenzie, C. / Green, S.R. / Huggett, M. / Barros, D. / Wyatt, P.G. / Ray, P.C. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 5oip.cif.gz | 127.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb5oip.ent.gz | 98.4 KB | Display | PDB format |
| PDBx/mmJSON format | 5oip.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/oi/5oipftp://data.pdbj.org/pub/pdb/validation_reports/oi/5oip | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  5oicC  5oifC  5oilC  5oimC  5oinC  5oioC  5oiqC  5oirC  5oisC  5oitC C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| |||||||||
| Unit cell |
| |||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | Mass: 28524.754 Da / Num. of mol.: 1 / Mutation: T2A Source method: isolated from a genetically manipulated source Source: (gene. exp.) Mycobacterium tuberculosis (bacteria) / Strain: ATCC 25618 / H37Rv / Gene: inhA, Rv1484, MTCY277.05 / Production host: References: UniProt: P9WGR1, enoyl-[acyl-carrier-protein] reductase (NADH) |
|---|---|
| #2: Chemical | ChemComp-NAD /   Mass: 663.425 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C21H27N7O14P2 / Comment: NAD*YM Mass: 663.425 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C21H27N7O14P2 / Comment: NAD*YM |
| #3: Chemical | ChemComp-9WE / 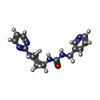  Mass: 312.370 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C16H20N6O / Feature type: SUBJECT OF INVESTIGATION Mass: 312.370 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C16H20N6O / Feature type: SUBJECT OF INVESTIGATION |
| #4: Chemical | ChemComp-PO4 /   Mass: 94.971 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: PO4 Mass: 94.971 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: PO4 |
| #5: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 304 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 304 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.52 Å3/Da / Density % sol: 65.05 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, sitting drop Details: 10% glycerol, 25% 1,2propanediol, 0.1M PO4. Cryo is well solution |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: Diamond  / Beamline: I04-1 / Wavelength: 0.9282 Å / Beamline: I04-1 / Wavelength: 0.9282 Å |
| Detector | Type: DECTRIS PILATUS3 6M / Detector: PIXEL / Date: Jul 22, 2016 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9282 Å / Relative weight: 1 |
| Reflection | Resolution: 1.71→41.34 Å / Num. obs: 45004 / % possible obs: 99.9 % / Redundancy: 19.2 % / Biso Wilson estimate: 31.31 Å2 / CC1/2: 1 / Rmerge(I) obs: 0.101 / Net I/σ(I): 19.6 |
| Reflection shell | Resolution: 1.71→1.75 Å / Redundancy: 17.1 % / Rmerge(I) obs: 2.876 / Mean I/σ(I) obs: 1.3 / Num. unique obs: 3248 / CC1/2: 0.514 / % possible all: 100 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 1.71→25.04 Å / Cor.coef. Fo:Fc: 0.965 / Cor.coef. Fo:Fc free: 0.966 / Rfactor Rfree error: 0 / SU R Cruickshank DPI: 0.072 / Cross valid method: THROUGHOUT / σ(F): 0 / SU R Blow DPI: 0.078 / SU Rfree Blow DPI: 0.073 / SU Rfree Cruickshank DPI: 0.069
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 36.29 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.2 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: 1 / Resolution: 1.71→25.04 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.71→1.75 Å / Rfactor Rfree error: 0 / Total num. of bins used: 20
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Origin x: 45.8577 Å / Origin y: 48.705 Å / Origin z: -88.9066 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group | Selection details: { A|* } |
