Mass: 18.015 Da / Num. of mol.: 141 / Source method: isolated from a natural source / Formula: H2O
-
Details
Sequence details
HEXAHISTIDINE-TAG WITH THROMBIN CLEAVAGE AT THE N-TERMINUS
-
Experimental details
-
Experiment
Experiment
Method: X-RAY DIFFRACTION / Number of used crystals: 1
-
Sample preparation
Crystal
Density Matthews: 2.93 Å3/Da / Density % sol: 58.1 % / Description: NONE
Crystal grow
Temperature: 281 K / Method: vapor diffusion, sitting drop / pH: 8 Details: PROTEIN 6.9 MG/ML 1 MM AMPCPP, 1 MM INHIBITOR, 2 MM MAGNESIUM CHLORIDE, AMMONIUM NITRATE 0.275 M, PEG3350 22.2% W/V, SITTING DROP, 281 K
Resolution: 1.56→42.33 Å / Cor.coef. Fo:Fc: 0.967 / Cor.coef. Fo:Fc free: 0.961 / SU B: 1.162 / SU ML: 0.042 / Cross valid method: THROUGHOUT / ESU R: 0.067 / ESU R Free: 0.067 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. U VALUES REFINED INDIVIDUALLY. AN ALTERNATE CONFORMATION OF THE LOOP 3 RESIDUE LYS86 IS SUGGESTED, THIS WAS NOT MODELLED DUE TO POOR ...Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. U VALUES REFINED INDIVIDUALLY. AN ALTERNATE CONFORMATION OF THE LOOP 3 RESIDUE LYS86 IS SUGGESTED, THIS WAS NOT MODELLED DUE TO POOR DENSITY. THE TERMINAL RESIDUE LYS158 IS SUGGESTED TO ADOPT MULTIPLE CONFORMATIONS. ONLY THE PREDOMINANT CONFORMATION IS MODELLED DUE TO ELSEWISE POOR DENSITY.
Rfactor
Num. reflection
% reflection
Selection details
Rfree
0.17816
1505
5.1 %
RANDOM
Rwork
0.15877
-
-
-
obs
0.15972
28270
99.68 %
-
Solvent computation
Ion probe radii: 0.7 Å / Shrinkage radii: 0.7 Å / VDW probe radii: 1.1 Å / Solvent model: MASK
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information
 STAPHYLOCOCCUS AUREUS (bacteria)
STAPHYLOCOCCUS AUREUS (bacteria) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads









 PDBj
PDBj



 Assembly
Assembly

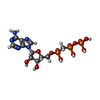


 Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg
Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg Mass: 505.208 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C11H18N5O12P3 / Comment: AMP-CPP, energy-carrying molecule analogue*YM
Mass: 505.208 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C11H18N5O12P3 / Comment: AMP-CPP, energy-carrying molecule analogue*YM Mass: 62.005 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: NO3
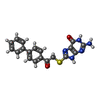
Mass: 62.005 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: NO3 Mass: 377.420 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C19H15N5O2S
Mass: 377.420 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C19H15N5O2S Sample preparation
Sample preparation / Beamline: MX2 / Wavelength: 0.9537
/ Beamline: MX2 / Wavelength: 0.9537  Processing
Processing