 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3qxo | ||||||
|---|---|---|---|---|---|---|---|
| Title | CDK2 in complex with inhibitor KVR-1-84 | ||||||
 Components Components | Cyclin-dependent kinase 2 | ||||||
 Keywords Keywords | TRANSFERASE/TRANSFERASE INHIBITOR / protein kinase / inhibitor / TRANSFERASE-TRANSFERASE INHIBITOR complex | ||||||
| Function / homology |  Function and homology information Function and homology informationcyclin A1-CDK2 complex / cyclin E2-CDK2 complex / cyclin E1-CDK2 complex / cyclin A2-CDK2 complex / positive regulation of DNA-templated DNA replication initiation / G2 Phase / Y chromosome / cyclin-dependent protein kinase activity / regulation of heterochromatin organization / Phosphorylation of proteins involved in G1/S transition by active Cyclin E:Cdk2 complexes ...cyclin A1-CDK2 complex / cyclin E2-CDK2 complex / cyclin E1-CDK2 complex / cyclin A2-CDK2 complex / positive regulation of DNA-templated DNA replication initiation / G2 Phase / Y chromosome / cyclin-dependent protein kinase activity / regulation of heterochromatin organization / Phosphorylation of proteins involved in G1/S transition by active Cyclin E:Cdk2 complexes / positive regulation of heterochromatin formation / p53-Dependent G1 DNA Damage Response / X chromosome / PTK6 Regulates Cell Cycle / regulation of anaphase-promoting complex-dependent catabolic process / Defective binding of RB1 mutants to E2F1,(E2F2, E2F3) / Regulation of APC/C activators between G1/S and early anaphase / centriole replication / telomere maintenance in response to DNA damage / centrosome duplication / G0 and Early G1 / Telomere Extension By Telomerase / Activation of the pre-replicative complex / cyclin-dependent kinase / cyclin-dependent protein serine/threonine kinase activity / TP53 Regulates Transcription of Genes Involved in G1 Cell Cycle Arrest / Regulation of MITF-M-dependent genes involved in cell cycle and proliferation / Activation of ATR in response to replication stress / Cajal body / Cyclin E associated events during G1/S transition / Cyclin A:Cdk2-associated events at S phase entry / cyclin-dependent protein kinase holoenzyme complex / Cyclin A/B1/B2 associated events during G2/M transition / regulation of G2/M transition of mitotic cell cycle / condensed chromosome / mitotic G1 DNA damage checkpoint signaling / negative regulation of protein localization to chromatin / cellular response to nitric oxide / post-translational protein modification / regulation of mitotic cell cycle / cyclin binding / positive regulation of DNA replication / peptidyl-serine phosphorylation / male germ cell nucleus / meiotic cell cycle / potassium ion transport / G1/S transition of mitotic cell cycle / DNA Damage/Telomere Stress Induced Senescence / Meiotic recombination / CDK-mediated phosphorylation and removal of Cdc6 / G2/M transition of mitotic cell cycle / SCF(Skp2)-mediated degradation of p27/p21 / Transcriptional regulation of granulopoiesis / Orc1 removal from chromatin / Cyclin D associated events in G1 / cellular senescence / Regulation of TP53 Degradation / nuclear envelope / Factors involved in megakaryocyte development and platelet production / Processing of DNA double-strand break ends / regulation of gene expression / Senescence-Associated Secretory Phenotype (SASP) / transcription regulator complex / Regulation of TP53 Activity through Phosphorylation / protein phosphorylation / Ras protein signal transduction / chromosome, telomeric region / DNA replication / endosome / chromatin remodeling / protein domain specific binding / protein serine kinase activity / cell division / DNA repair / protein serine/threonine kinase activity / positive regulation of cell population proliferation / DNA-templated transcription / centrosome / positive regulation of DNA-templated transcription / magnesium ion binding / negative regulation of transcription by RNA polymerase II / signal transduction / nucleoplasm / ATP binding / nucleus / cytoplasm / cytosol Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.75 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.75 Å | ||||||
 Authors Authors | Betzi, S. / Alam, R. / Han, H. / Becker, A. / Schonbrunn, E. | ||||||
 Citation Citation | Journal: To be Published Title: Structure-guided optimization of novel CDK2 inhibitors discovered by high-throughput screening Authors: Schonbrunn, E. / Becker, A. / Betzi, S. / Alam, R. / Han, H. / Rawle, F. / Katta, V. / Jakkaraj, J. / Chakrasali, R. / Neelam, S. / Hook, D. / Tash, J. / Georg, G. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3qxo.cif.gz | 79 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3qxo.ent.gz | 57.5 KB | Display | PDB format |
| PDBx/mmJSON format | 3qxo.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/qx/3qxoftp://data.pdbj.org/pub/pdb/validation_reports/qx/3qxo | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  3ql8C  3qqfC  3qqgC  3qqhC  3qqjC  3qqlC  3qrtC  3qruC  3qwjC  3qwkC  3qx2C  3qx4C  3qzfC  3qzgC  3qzhC  3qziC  3r1qC  3r1sC  3r1yC  3r28C  3r6xC  3r71C  3r73C  3r7eC  3r7iC  3r7uC  3r7vC  3r7yC  3r83C  3r8lC  3r8mC  3r8pC  3raiC  3rm6C  3rm7C  3royC  3rpoC  1pw2S C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |






















































-Links
 PDBj
PDBj









- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 34761.348 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: CDK2 / Plasmid: pGEX6P-1 / Production host:  |
|---|---|
| #2: Chemical | ChemComp-EDO /   Mass: 62.068 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6O2 Mass: 62.068 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6O2 |
| #3: Chemical | ChemComp-X65 / 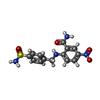  Mass: 350.350 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C14H14N4O5S Mass: 350.350 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C14H14N4O5S |
| #4: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 211 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 211 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.01 Å3/Da / Density % sol: 38.73 % |
|---|---|
| Crystal grow | Temperature: 291 K / Method: vapor diffusion, hanging drop / pH: 7.5 Details: 5 mg/mL CDK2 protein, 3 mM inhibitor, 15% v/v PEG3350, 50 mM HEPES/NaOH, 50 mM Na/K phosphate, pH 7.5, VAPOR DIFFUSION, HANGING DROP, temperature 291K |
-Data collection
| Diffraction | Mean temperature: 93 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU MICROMAX-007 HF / Wavelength: 1.54178 |
| Detector | Type: RIGAKU SATURN 944+ / Detector: CCD / Date: Jan 28, 2009 / Details: MIRRORS |
| Radiation | Monochromator: MIRRORS / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.54178 Å / Relative weight: 1 |
| Reflection | Resolution: 1.75→20 Å / Num. all: 28872 / Num. obs: 28872 / % possible obs: 99.9 % / Observed criterion σ(F): 0 / Observed criterion σ(I): -3 / Redundancy: 6.96 % / Biso Wilson estimate: 18.2 Å2 / Rmerge(I) obs: 0.046 / Net I/σ(I): 27.01 |
| Reflection shell | Resolution: 1.75→1.8 Å / Redundancy: 6.69 % / Rmerge(I) obs: 0.241 / Mean I/σ(I) obs: 9.25 / Num. unique all: 2314 / % possible all: 99.9 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1PW2 Resolution: 1.75→18.77 Å / Rfactor Rfree error: 0.007 / Data cutoff high absF: 1443524.86 / Data cutoff low absF: 0 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0 / Stereochemistry target values: Engh & Huber / Details: BULK SOLVENT MODEL USED
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 55.6421 Å2 / ksol: 0.4 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 26.6 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.75→18.77 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints NCS | NCS model details: NONE | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.75→1.86 Å / Rfactor Rfree error: 0.022 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
|
