 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-3pcb: STRUCTURE OF PROTOCATECHUATE 3,4-DIOXYGENASE COMPLEXED WITH 3-HYD... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3pcb | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | STRUCTURE OF PROTOCATECHUATE 3,4-DIOXYGENASE COMPLEXED WITH 3-HYDROXYBENZOATE | ||||||||||||
 Components Components |
| ||||||||||||
 Keywords Keywords | DIOXYGENASE / IRON / NONHEME / METALLOPROTEIN / OXIDOREDUCTASE | ||||||||||||
| Function / homology |  Function and homology information Function and homology informationprotocatechuate 3,4-dioxygenase / protocatechuate 3,4-dioxygenase activity / 3,4-dihydroxybenzoate catabolic process / beta-ketoadipate pathway / ferric iron binding Similarity search - Function | ||||||||||||
| Biological species |  Pseudomonas putida (bacteria) Pseudomonas putida (bacteria) | ||||||||||||
| Method |  X-RAY DIFFRACTION / NATIVE MODEL PHASES / Resolution: 2.19 Å X-RAY DIFFRACTION / NATIVE MODEL PHASES / Resolution: 2.19 Å | ||||||||||||
 Authors Authors | Elango, N. / Orville, A.M. / Lipscomb, J.D. / Ohlendorf, D.H. | ||||||||||||
 Citation Citation | Journal: Biochemistry / Year: 1997 Title: Structures of competitive inhibitor complexes of protocatechuate 3,4-dioxygenase: multiple exogenous ligand binding orientations within the active site. Authors: Orville, A.M. / Elango, N. / Lipscomb, J.D. / Ohlendorf, D.H. #1: Journal: J.Mol.Biol. / Year: 1994Title: Structure of Protocatechuate 3,4-Dioxygenase from Pseudomonas Aeruginosa at 2.15 A Resolution Authors: Ohlendorf, D.H. / Orville, A.M. / Lipscomb, J.D. #2: Journal: Nature / Year: 1988Title: Structure and Assembly of Protocatechuate 3,4-Dioxygenase Authors: Ohlendorf, D.H. / Lipscomb, J.D. / Weber, P.C. #3: Journal: J.Mol.Biol. / Year: 1987Title: Determination of the Quaternary Structure of Protocatechuate 3,4-Dioxygenase from Pseudomonas Aeruginosa Authors: Ohlendorf, D.H. / Weber, P.C. / Lipscomb, J.D. | ||||||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3pcb.cif.gz | 544.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3pcb.ent.gz | 440 KB | Display | PDB format |
| PDBx/mmJSON format | 3pcb.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/pc/3pcbftp://data.pdbj.org/pub/pdb/validation_reports/pc/3pcb | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  3pccC  3pceC  3pcfC  3pcgC  3pchC  3pciC C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||||||||||||
| Unit cell |
| ||||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS oper:
| ||||||||||||||||||
| Details | THE ACTIVE FORM OF THE ENZYME IS THE WHOLE UNIT CELL CONTENT. IT CONSISTS OF 12 PROTOMERS. EACH PROTOMER IS MADE OF TWO POLYPEPTIDE CHAINS AND ONE FE(III). THE ASYMMETRIC UNIT CONSISTS OF 6 PROTOMERS. THEY ARE: CHAINS (A, M), CHAINS (B, N), CHAINS (C, O), CHAINS (D, P), CHAINS (E, Q), CHAINS (F, R). EACH PROTOMER CONTAINS AN ACTIVE SITE WITH FE(III) AS THE CENTER AND A VESTIGIAL SITE. THE ACTIVE SITES ARE NAMED: ACA, ACB, ACC, ACD, ACE, ACF. THE VESTIGIAL SITES ARE NAMED VEA, VEB, VEC, VED, VEE AND VEF. THE CHAIN IDENTIFIERS HAVE BEEN ASSIGNED AS FOLLOWS: ALPHA BETA WATERS A M G B N H C O I D P J E Q K F R L |
-Components
| #1: Protein | Mass: 22278.812 Da / Num. of mol.: 6 / Source method: isolated from a natural source / Details: ENTRY CONTAINS ALPHA/BETA 6-MER / Source: (natural) Pseudomonas putida (bacteria)References: UniProt: P00436, protocatechuate 3,4-dioxygenase #2: Protein | Mass: 26772.404 Da / Num. of mol.: 6 / Source method: isolated from a natural source / Details: ENTRY CONTAINS ALPHA/BETA 6-MER / Source: (natural) Pseudomonas putida (bacteria)References: UniProt: P00437, protocatechuate 3,4-dioxygenase #3: Chemical | ChemComp-FE /   Mass: 55.845 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: Fe Mass: 55.845 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: Fe#4: Chemical | ChemComp-3HB / 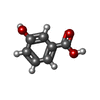  Mass: 138.121 Da / Num. of mol.: 12 / Source method: obtained synthetically / Formula: C7H6O3 Mass: 138.121 Da / Num. of mol.: 12 / Source method: obtained synthetically / Formula: C7H6O3#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 1416 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 1416 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.84 Å3/Da / Density % sol: 56.74 % | |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 8.4 / Details: pH 8.4 | |||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 20 ℃ / Method: interface diffusionDetails: or hanging drop vapor diffusion, Ohlendorf, D.H., (1994) J.Mol.Biol., 244, 586. | |||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 293 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RUH2R / Wavelength: 1.5418 |
| Detector | Type: SIEMENS / Detector: AREA DETECTOR / Date: Sep 3, 1994 / Details: COLLIMATOR |
| Radiation | Monochromator: GRAPHITE(002) / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2.19→15 Å / Num. obs: 132191 / % possible obs: 78.2 % / Observed criterion σ(I): 1 / Redundancy: 1.9 % / Rsym value: 0.06 / Net I/σ(I): 14.6 |
| Reflection shell | Resolution: 2.19→2.28 Å / Redundancy: 2 % / Mean I/σ(I) obs: 1.5 / % possible all: 36.5 |
| Reflection | *PLUS Num. measured all: 247730 / Rmerge(I) obs: 0.152 |
| Reflection shell | *PLUS % possible obs: 36.5 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: NATIVE MODEL PHASES / Resolution: 2.19→6 Å / σ(F): 1 Details: APART FROM BINDING THE INHIBITOR AT THE ACTIVE SITE OF EACH PROTOMER, ONE MOLECULE OF THE INHIBITOR CORRESPONDING TO EACH PROTOMER ALSO BINDS AT THE LOCAL THREE-FOLD SYMMETRY. THE ...Details: APART FROM BINDING THE INHIBITOR AT THE ACTIVE SITE OF EACH PROTOMER, ONE MOLECULE OF THE INHIBITOR CORRESPONDING TO EACH PROTOMER ALSO BINDS AT THE LOCAL THREE-FOLD SYMMETRY. THE NONSTANDARD UNIT CELL I 2 WAS CHOSEN OVER THE EQUIVALENT C 2 CELL BECAUSE OF THE CONVENIENCE OF HAVING A BETA ANGLE NEAR 90 DEGREES AND TO MAINTAIN CONSISTENCY WITH THE INITIAL CRYSTALLIZATION REPORT. TO CONVERT FROM FRACTIONAL I 2 TO FRACTIONAL C 2, USE THE FOLLOWING TRANSFORMATION: [ 1.0 ][ 0.0 ][ 0.0 ] (XFRAC_I2) (XFRAC_C2) [ 0.0 ][ 1.0 ][ 0.0 ] X (YFRAC_I2) = (YFRAC_C2) [ -1.0 ][ 0.0 ][ 1.0 ] (ZFRAC_I2) (ZFRAC_C2) THE ORTHOGONAL COORDINATE SYSTEM CHOSEN FOR THIS ENTRY IS THAT DEFINED BY THE LOCAL SYMMETRY OF THE COMPLEX (23).
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 25.76 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.19→6 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: PROLSQ / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Rfactor obs: 0.159 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Highest resolution: 2.2 Å / Lowest resolution: 2.4 Å / Rfactor Rwork: 0.211 |
