 ムービー
ムービー コントローラー
コントローラー


+ データを開く
データを開く

- 基本情報
基本情報
| 登録情報 |  | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| タイトル | Accurate computational design of genetically encoded 3D protein crystals | |||||||||
 マップデータ マップデータ | Sharpened | |||||||||
 試料 試料 |
| |||||||||
 キーワード キーワード | 3D crystals / nanocage / de novo design / rosetta / cryoEM / DE NOVO PROTEIN | |||||||||
| 生物種 |  | |||||||||
| 手法 | 単粒子再構成法 / クライオ電子顕微鏡法 / 解像度: 3.34 Å | |||||||||
 データ登録者 データ登録者 | Li Z / Borst AJ / Baker D | |||||||||
| 資金援助 |  米国, 1件 米国, 1件
| |||||||||
 引用 引用 | ジャーナル: Nat Mater / 年: 2023 タイトル: Accurate computational design of three-dimensional protein crystals. 著者: Zhe Li / Shunzhi Wang / Una Nattermann / Asim K Bera / Andrew J Borst / Muammer Y Yaman / Matthew J Bick / Erin C Yang / William Sheffler / Byeongdu Lee / Soenke Seifert / Greg L Hura / ...著者: Zhe Li / Shunzhi Wang / Una Nattermann / Asim K Bera / Andrew J Borst / Muammer Y Yaman / Matthew J Bick / Erin C Yang / William Sheffler / Byeongdu Lee / Soenke Seifert / Greg L Hura / Hannah Nguyen / Alex Kang / Radhika Dalal / Joshua M Lubner / Yang Hsia / Hugh Haddox / Alexis Courbet / Quinton Dowling / Marcos Miranda / Andrew Favor / Ali Etemadi / Natasha I Edman / Wei Yang / Connor Weidle / Banumathi Sankaran / Babak Negahdari / Michael B Ross / David S Ginger / David Baker /  要旨: Protein crystallization plays a central role in structural biology. Despite this, the process of crystallization remains poorly understood and highly empirical, with crystal contacts, lattice packing ...Protein crystallization plays a central role in structural biology. Despite this, the process of crystallization remains poorly understood and highly empirical, with crystal contacts, lattice packing arrangements and space group preferences being largely unpredictable. Programming protein crystallization through precisely engineered side-chain-side-chain interactions across protein-protein interfaces is an outstanding challenge. Here we develop a general computational approach for designing three-dimensional protein crystals with prespecified lattice architectures at atomic accuracy that hierarchically constrains the overall number of degrees of freedom of the system. We design three pairs of oligomers that can be individually purified, and upon mixing, spontaneously self-assemble into >100 µm three-dimensional crystals. The structures of these crystals are nearly identical to the computational design models, closely corresponding in both overall architecture and the specific protein-protein interactions. The dimensions of the crystal unit cell can be systematically redesigned while retaining the space group symmetry and overall architecture, and the crystals are extremely porous and highly stable. Our approach enables the computational design of protein crystals with high accuracy, and the designed protein crystals, which have both structural and assembly information encoded in their primary sequences, provide a powerful platform for biological materials engineering. | |||||||||
| 履歴 |
|
- 構造の表示
構造の表示
| 添付画像 |
|---|

- ダウンロードとリンク
ダウンロードとリンク
-EMDBアーカイブ
| マップデータ | emd_27031.map.gz | 117.5 MB |  EMDBマップデータ形式 EMDBマップデータ形式 | |
|---|---|---|---|---|
| ヘッダ (付随情報) | emd-27031-v30.xmlemd-27031.xml | 19.1 KB 19.1 KB | 表示 表示 | EMDBヘッダ |
| 画像 |  emd_27031.png emd_27031.png | 72.9 KB | ||
| Filedesc metadata | emd-27031.cif.gz | 5.5 KB | ||
| その他 | emd_27031_additional_1.map.gzemd_27031_half_map_1.map.gzemd_27031_half_map_2.map.gz | 58.9 MB 114.4 MB 114.4 MB | ||
| アーカイブディレクトリ |  http://ftp.pdbj.org/pub/emdb/structures/EMD-27031ftp://ftp.pdbj.org/pub/emdb/structures/EMD-27031 http://ftp.pdbj.org/pub/emdb/structures/EMD-27031ftp://ftp.pdbj.org/pub/emdb/structures/EMD-27031 | HTTPS FTP |
-検証レポート
| 文書・要旨 | emd_27031_validation.pdf.gz | 768.8 KB | 表示 | EMDB検証レポート |
|---|---|---|---|---|
| 文書・詳細版 | emd_27031_full_validation.pdf.gz | 768.4 KB | 表示 | |
| XML形式データ | emd_27031_validation.xml.gz | 14 KB | 表示 | |
| CIF形式データ | emd_27031_validation.cif.gz | 16.3 KB | 表示 | |
| アーカイブディレクトリ | https://ftp.pdbj.org/pub/emdb/validation_reports/EMD-27031ftp://ftp.pdbj.org/pub/emdb/validation_reports/EMD-27031 | HTTPS FTP |
-関連構造データ












-リンク
| EMDBのページ | EMDB (EBI/PDBe) / EMDataResource |
|---|
-マップ
| ファイル | ダウンロード / ファイル: emd_27031.map.gz / 形式: CCP4 / 大きさ: 125 MB / タイプ: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 注釈 | Sharpened | ||||||||||||||||||||
| ボクセルのサイズ | X=Y=Z: 1.0288 Å | ||||||||||||||||||||
| 密度 |
| ||||||||||||||||||||
| 対称性 | 空間群: 1 | ||||||||||||||||||||
| 詳細 | EMDB XML:
|
-添付データ
-追加マップ: Unsharpened
| ファイル | emd_27031_additional_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 注釈 | Unsharpened | ||||||||||||
| 投影像・断面図 |
| ||||||||||||
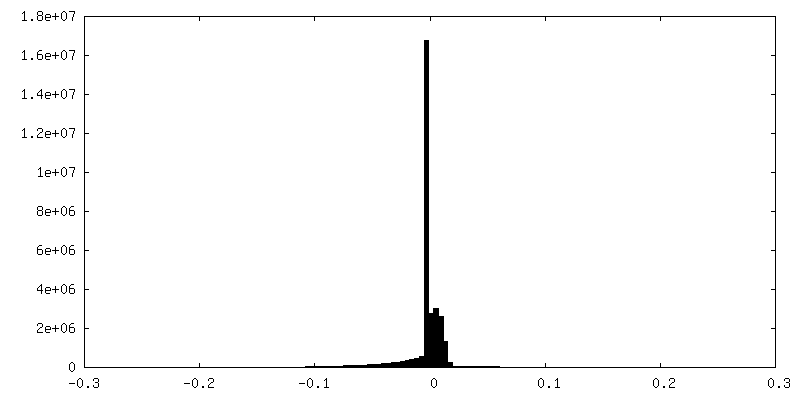
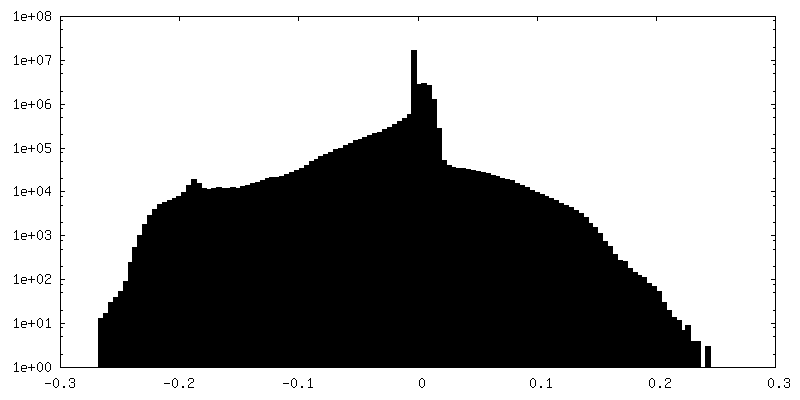
| 密度ヒストグラム |
 Z
Z Y
Y X
X







-ハーフマップ: Half A
| ファイル | emd_27031_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 注釈 | Half A | ||||||||||||
| 投影像・断面図 |
| ||||||||||||
| 密度ヒストグラム |








-ハーフマップ: Half B
| ファイル | emd_27031_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 注釈 | Half B | ||||||||||||
| 投影像・断面図 |
| ||||||||||||
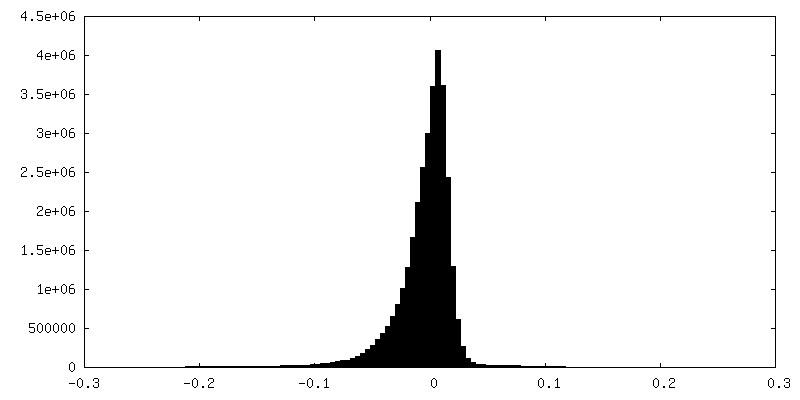
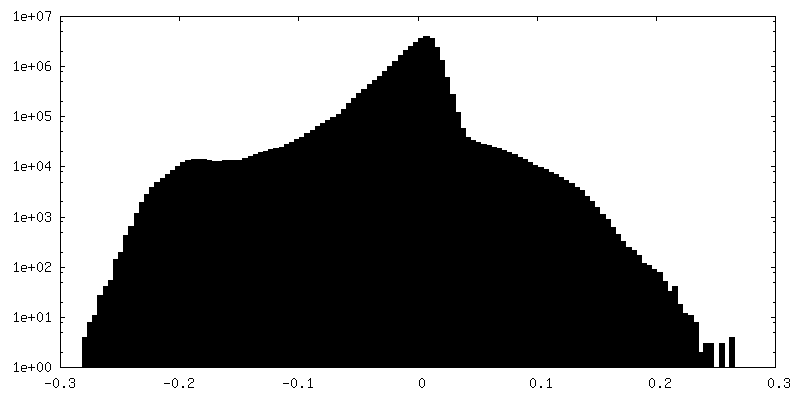
| 密度ヒストグラム |






- 試料の構成要素
試料の構成要素
-全体 : T32-15
| 全体 | 名称: T32-15 |
|---|---|
| 要素 |
|
-超分子 #1: T32-15
| 超分子 | 名称: T32-15 / タイプ: complex / ID: 1 / 親要素: 0 / 含まれる分子: all |
|---|---|
| 由来(天然) | 生物種: |
-分子 #1: T32-15-1
| 分子 | 名称: T32-15-1 / タイプ: protein_or_peptide / ID: 1 / コピー数: 12 / 光学異性体: LEVO |
|---|---|
| 由来(天然) | 生物種: synthetic construct (人工物) |
| 分子量 | 理論値: 49.525008 KDa |
| 組換発現 | 生物種: |
| 配列 | 文字列: DEAEEKARRV AEKVERLKRS GTSEDEIAEE VAREISEVIR TLKESGSSYE VIAEIVARIV AEIVEALKRS GTSEDEIAEI VARVISEVI RTLKESGSSY EVIAEIVARI VAEIVEALKR SGTSEDEIAE IVARVISEVI RTLKESGSSY EVIAEIVARI V AEIVEALK ...文字列: DEAEEKARRV AEKVERLKRS GTSEDEIAEE VAREISEVIR TLKESGSSYE VIAEIVARIV AEIVEALKRS GTSEDEIAEI VARVISEVI RTLKESGSSY EVIAEIVARI VAEIVEALKR SGTSEDEIAE IVARVISEVI RTLKESGSSY EVIAEIVARI V AEIVEALK RSGTSEDEIA EIVARVISEV IRTLKESGSS AEVIAEIVAR IVAEIVEALK RSGTSEDEIA EIVARVISEV IR TLKESGS SSILIALIVA RIVAEIVEAL KRSGTSEDEI AEIVARVISE VIRTLKESGS SYEIIALIVA MIVAEIVRAL LRS GTSEEE IAKIVARVMN EVLRTLRESG SDFEVIREIL RLILAAIRAA LQKGGVSEDE IMRIEIKILL MLLRLSTAEL ERAT RSLKA ITEELKKNPS EDALVEHNRA IVEHNRIIVF NNILIALVLE AIVRAI |
-分子 #2: T32-15-2
| 分子 | 名称: T32-15-2 / タイプ: protein_or_peptide / ID: 2 / コピー数: 12 / 光学異性体: LEVO |
|---|---|
| 由来(天然) | 生物種: synthetic construct (人工物) |
| 分子量 | 理論値: 9.066595 KDa |
| 組換発現 | 生物種: |
| 配列 | 文字列: TRTEIIRELE RSLREQEELA KRLMELLLKL LRLQMTGSSD EDVRRLMLRI IELVEEIEEL AREQKYLVEE LKRQ |
-実験情報
-構造解析
| 手法 | クライオ電子顕微鏡法 |
|---|---|
 解析 解析 | 単粒子再構成法 |
| 試料の集合状態 | particle |
-試料調製
| 濃度 | 5.3 mg/mL |
|---|---|
| 緩衝液 | pH: 8 |
| グリッド | モデル: C-flat-1.2/1.3 / 材質: COPPER / メッシュ: 400 / 支持フィルム - 材質: CARBON / 支持フィルム - トポロジー: HOLEY |
| 凍結 | 凍結剤: ETHANE |
- 電子顕微鏡法
電子顕微鏡法
| 顕微鏡 | FEI TITAN KRIOS |
|---|---|
| 撮影 | フィルム・検出器のモデル: GATAN K3 (6k x 4k) / 平均電子線量: 50.0 e/Å2 |
| 電子線 | 加速電圧: 300 kV / 電子線源:  FIELD EMISSION GUN FIELD EMISSION GUN |
| 電子光学系 | 照射モード: FLOOD BEAM / 撮影モード: BRIGHT FIELD / 最大 デフォーカス(公称値): 2.2 µm / 最小 デフォーカス(公称値): 0.8 µm |
| 実験機器 |  モデル: Titan Krios / 画像提供: FEI Company |
-画像解析
| 初期モデル | モデルのタイプ: NONE / 詳細: Ab initio |
|---|---|
| 最終 再構成 | 解像度のタイプ: BY AUTHOR / 解像度: 3.34 Å / 解像度の算出法: FSC 0.143 CUT-OFF / 使用した粒子像数: 511464 |
| 初期 角度割当 | タイプ: NOT APPLICABLE / ソフトウェア - 名称: cryoSPARC (ver. 3.2) |
| 最終 角度割当 | タイプ: MAXIMUM LIKELIHOOD / ソフトウェア - 名称: cryoSPARC (ver. 3.2) |
