 ムービー
ムービー コントローラー
コントローラー


+ データを開く
データを開く

- 基本情報
基本情報
| 登録情報 | データベース: EMDB / ID: EMD-2257 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| タイトル | negative stain single-particle reconstruction of conformation X of the Ltn1 E3 ubiquitin Ligase | |||||||||
 マップデータ マップデータ | conformational snapshot X of Ltn1 | |||||||||
 試料 試料 |
| |||||||||
 キーワード キーワード | conformational heterogeneity / Ltn1/Listerin / RING E3 ubiquitin ligase / translational surveillance / neurodegenerative disease | |||||||||
| 生物種 |  | |||||||||
| 手法 | 単粒子再構成法 / ネガティブ染色法 / 解像度: 40.6 Å | |||||||||
 データ登録者 データ登録者 | Lyumkis D / Doamekpor SK / Bengtson MH / Lee JW / Toro TB / Petroski MD / Lima CD / Potter CS / Carragher B / Joazeiro CAP | |||||||||
 引用 引用 | ジャーナル: Proc Natl Acad Sci U S A / 年: 2013 タイトル: Single-particle EM reveals extensive conformational variability of the Ltn1 E3 ligase. 著者: Dmitry Lyumkis / Selom K Doamekpor / Mario H Bengtson / Joong-Won Lee / Tasha B Toro / Matthew D Petroski / Christopher D Lima / Clinton S Potter / Bridget Carragher / Claudio A P Joazeiro /  要旨: Ltn1 is a 180-kDa E3 ubiquitin ligase that associates with ribosomes and marks certain aberrant, translationally arrested nascent polypeptide chains for proteasomal degradation. In addition to its ...Ltn1 is a 180-kDa E3 ubiquitin ligase that associates with ribosomes and marks certain aberrant, translationally arrested nascent polypeptide chains for proteasomal degradation. In addition to its evolutionarily conserved large size, Ltn1 is characterized by the presence of a conserved N terminus, HEAT/ARM repeats predicted to comprise the majority of the protein, and a C-terminal catalytic RING domain, although the protein's exact structure is unknown. We used numerous single-particle EM strategies to characterize Ltn1's structure based on negative stain and vitreous ice data. Two-dimensional classifications and subsequent 3D reconstructions of electron density maps show that Ltn1 has an elongated form and presents a continuum of conformational states about two flexible hinge regions, whereas its overall architecture is reminiscent of multisubunit cullin-RING ubiquitin ligase complexes. We propose a model of Ltn1 function based on its conformational variability and flexibility that describes how these features may play a role in cotranslational protein quality control. | |||||||||
| 履歴 |
|
- 構造の表示
構造の表示
| ムービー |
 ムービービューア ムービービューア |
|---|---|
| 構造ビューア | EMマップ: SurfViewMolmilJmol/JSmol |
| 添付画像 |

- ダウンロードとリンク
ダウンロードとリンク
-EMDBアーカイブ
| マップデータ | emd_2257.map.gz | 14.1 MB | EMDBマップデータ形式 | |
|---|---|---|---|---|
| ヘッダ (付随情報) | emd-2257-v30.xmlemd-2257.xml | 13.8 KB 13.8 KB | 表示 表示 | EMDBヘッダ |
| 画像 | 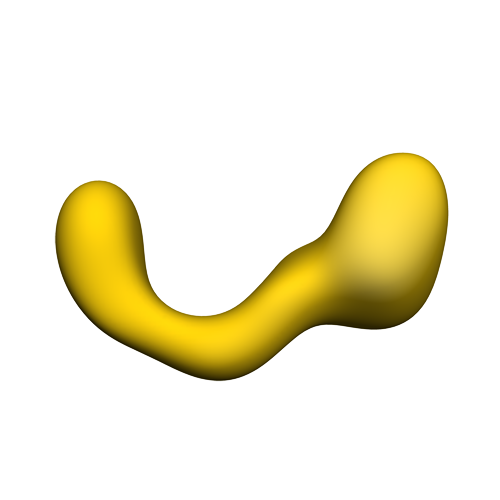 EMD-2257.png EMD-2257.png | 49.9 KB | ||
| アーカイブディレクトリ |  http://ftp.pdbj.org/pub/emdb/structures/EMD-2257ftp://ftp.pdbj.org/pub/emdb/structures/EMD-2257 http://ftp.pdbj.org/pub/emdb/structures/EMD-2257ftp://ftp.pdbj.org/pub/emdb/structures/EMD-2257 | HTTPS FTP |
-検証レポート
| 文書・要旨 | emd_2257_validation.pdf.gz | 193 KB | 表示 | EMDB検証レポート |
|---|---|---|---|---|
| 文書・詳細版 | emd_2257_full_validation.pdf.gz | 192.2 KB | 表示 | |
| XML形式データ | emd_2257_validation.xml.gz | 5.8 KB | 表示 | |
| アーカイブディレクトリ | https://ftp.pdbj.org/pub/emdb/validation_reports/EMD-2257ftp://ftp.pdbj.org/pub/emdb/validation_reports/EMD-2257 | HTTPS FTP |
-関連構造データ
| 関連構造データ |  2248C  2249C  2250C  2251C  2252C  2253C  2254C  2255C  2256C  2258C  2259C  2260C  2261C  2262C  2263C  2264C  2265C  2266C  2267C  2268C  2269C C: 同じ文献を引用 ( |
|---|---|
| 類似構造データ |


-リンク
| EMDBのページ | EMDB (EBI/PDBe) / EMDataResource |
|---|
-マップ
| ファイル | ダウンロード / ファイル: emd_2257.map.gz / 形式: CCP4 / 大きさ: 15.3 MB / タイプ: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 注釈 | conformational snapshot X of Ltn1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 投影像・断面図 | 画像のコントロール
画像は Spider により作成 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ボクセルのサイズ | X=Y=Z: 2.18 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
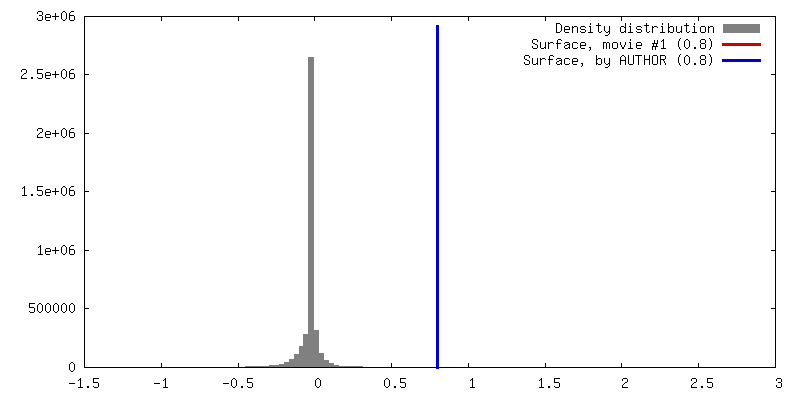
| 密度 |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 対称性 | 空間群: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 詳細 | EMDB XML:
CCP4マップ ヘッダ情報:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)




















-添付データ
- 試料の構成要素
試料の構成要素
-全体 : snapshot X of Ltn1 conformer
| 全体 | 名称: snapshot X of Ltn1 conformer |
|---|---|
| 要素 |
|
-超分子 #1000: snapshot X of Ltn1 conformer
| 超分子 | 名称: snapshot X of Ltn1 conformer / タイプ: sample / ID: 1000 / Number unique components: 1 |
|---|---|
| 分子量 | 実験値: 180 MDa / 理論値: 180 MDa / 手法: SDS-PAGE |
-分子 #1: Ltn1
| 分子 | 名称: Ltn1 / タイプ: protein_or_peptide / ID: 1 / Name.synonym: Listerin / コピー数: 1 / 集合状態: monomer / 組換発現: Yes |
|---|---|
| 由来(天然) | 生物種: |
| 分子量 | 実験値: 180 MDa / 理論値: 180 MDa |
| 組換発現 | 生物種:  |
-実験情報
-構造解析
| 手法 | ネガティブ染色法 |
|---|---|
 解析 解析 | 単粒子再構成法 |
| 試料の集合状態 | particle |
-試料調製
| 濃度 | 0.01 mg/mL |
|---|---|
| 緩衝液 | pH: 8 / 詳細: 190 mM NaCl, 20 mM Tris, 1mM BME |
| 染色 | タイプ: NEGATIVE 詳細: 3 microliters of sample at a concentration of 0.01 mg/mL was applied to a C-flat grid with 2-micron-diameter holes overlaid by thin 1.5 nm continuous carbon. The specimen was stained with 2% ...詳細: 3 microliters of sample at a concentration of 0.01 mg/mL was applied to a C-flat grid with 2-micron-diameter holes overlaid by thin 1.5 nm continuous carbon. The specimen was stained with 2% uranyl formate 3 times, then let air-dry. |
| グリッド | 詳細: c-flat grids overlaid with thin (~1.5 nm) carbon |
| 凍結 | 凍結剤: NONE / 装置: OTHER |
- 電子顕微鏡法
電子顕微鏡法
| 顕微鏡 | FEI TECNAI F20 |
|---|---|
| 温度 | 平均: 298 K |
| 日付 | 2011年3月23日 |
| 撮影 | カテゴリ: CCD フィルム・検出器のモデル: GATAN ULTRASCAN 4000 (4k x 4k) 実像数: 489 / 平均電子線量: 15 e/Å2 |
| Tilt angle max | 0 |
| 電子線 | 加速電圧: 120 kV / 電子線源:  FIELD EMISSION GUN FIELD EMISSION GUN |
| 電子光学系 | 倍率(補正後): 50000 / 照射モード: FLOOD BEAM / 撮影モード: BRIGHT FIELD / Cs: 2 mm / 最大 デフォーカス(公称値): 2.6 µm / 最小 デフォーカス(公称値): 1.2 µm / 倍率(公称値): 50000 |
| 試料ステージ | 試料ホルダー: single-tilt room temperature / 試料ホルダーモデル: SIDE ENTRY, EUCENTRIC / Tilt angle min: -55 |
| 実験機器 |  モデル: Tecnai F20 / 画像提供: FEI Company |
-画像解析
| 詳細 | random-conical tilt reconstruction |
|---|---|
| CTF補正 | 詳細: each micrograph |
| 最終 再構成 | 想定した対称性 - 点群: C1 (非対称) / アルゴリズム: OTHER / 解像度のタイプ: BY AUTHOR / 解像度: 40.6 Å / 解像度の算出法: FSC 0.5 CUT-OFF / ソフトウェア - 名称: Spider, Appion / 使用した粒子像数: 946 |
| 最終 2次元分類 | クラス数: 1 |
