 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1pjx | ||||||
|---|---|---|---|---|---|---|---|
| Title | 0.85 ANGSTROM STRUCTURE OF SQUID GANGLION DFPASE | ||||||
 Components Components | DIISOPROPYLFLUOROPHOSPHATASE | ||||||
 Keywords Keywords | HYDROLASE / PHOSPHOTRIESTERASE (PTE) / NITROGEN-CALCIUM COORDINATION / BETA-PROPELLER / BOND-LENGTH AND BOND-ANGLE RESTRAINTS / TORSION ANGLES / ROTAMER CLASSIFICATION | ||||||
| Function / homology |  Function and homology information Function and homology informationdiisopropyl-fluorophosphatase / diisopropyl-fluorophosphatase activity / calcium ion binding Similarity search - Function | ||||||
| Biological species |  Loligo vulgaris (squid) Loligo vulgaris (squid) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 0.85 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 0.85 Å | ||||||
 Authors Authors | Koepke, J. / Rueterjans, H. / Luecke, C. / Fritzsch, G. | ||||||
 Citation Citation | Journal: Acta Crystallogr.,Sect.D / Year: 2003 Title: Statistical analysis of crystallographic data obtained from squid ganglion DFPase at 0.85 A resolution. Authors: Koepke, J. / Scharff, E.I. / Lucke, C. / Ruterjans, H. / Fritzsch, G. #1: Journal: Acta Crystallogr.,Sect.D / Year: 2001Title: Crystallization and Preliminary X-ray Crystallographic Analysis of DFPase from Loligo Vulgaris Authors: Scharff, E.I. / Luecke, C. / Fritzsch, G. / Koepke, J. / Hartleib, J. / Dierl, S. / Rueterjans, H. #2: Journal: Structure / Year: 2001Title: Crystal Structure of Diisopropylfluorophosphatase from Loligo Vulgaris Authors: Scharff, E.I. / Koepke, J. / Fritzsch, G. / Luecke, C. / Rueterjans, H. #3: Journal: Acta Crystallogr.,Sect.D / Year: 2002Title: Atomic Resolution Crystal Structure of Squid Ganglion DFPase Authors: Koepke, J. / Scharff, E.I. / Luecke, C. / Rueterjans, H. / Fritzsch, G. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1pjx.cif.gz | 101.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1pjx.ent.gz | 74 KB | Display | PDB format |
| PDBx/mmJSON format | 1pjx.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/pj/1pjxftp://data.pdbj.org/pub/pdb/validation_reports/pj/1pjx | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1e1aS S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj



- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
-Protein , 1 types, 1 molecules A
| #1: Protein | Mass: 35120.711 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Loligo vulgaris (squid) / Organ: HEAD GANGLION / Plasmid: PKKHISND / Species (production host): Escherichia coli / Production host:  |
|---|
-Non-polymers , 10 types, 503 molecules 
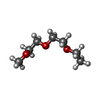




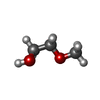



| #2: Chemical |  Mass: 40.078 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Ca Mass: 40.078 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Ca#3: Chemical | ChemComp-ME2 / |  Mass: 148.200 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C7H16O3 Mass: 148.200 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C7H16O3#4: Chemical |  Mass: 195.237 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C6H13NO4S / Comment: pH buffer*YM Mass: 195.237 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C6H13NO4S / Comment: pH buffer*YM#5: Chemical | ChemComp-EDO /  Mass: 62.068 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: C2H6O2 Mass: 62.068 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: C2H6O2#6: Chemical |  Mass: 150.173 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C6H14O4 Mass: 150.173 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C6H14O4#7: Chemical |  Mass: 90.121 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C4H10O2 Mass: 90.121 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C4H10O2#8: Chemical |  Mass: 76.094 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C3H8O2 Mass: 76.094 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C3H8O2#9: Chemical |  Mass: 92.094 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C3H8O3#10: Chemical |  Mass: 106.120 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C4H10O3 Mass: 106.120 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C4H10O3#11: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 479 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Details
| Has protein modification | N |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 4 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 1.79 Å3/Da / Density % sol: 43.34 % |
|---|---|
| Crystal grow | pH: 6.5 / Details: pH 6.5 |
-Data collection
| Diffraction | Mean temperature: 120 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: EMBL/DESY, HAMBURG  / Beamline: BW7B / Wavelength: 0.84 / Beamline: BW7B / Wavelength: 0.84 |
| Detector | Type: MAR scanner 345 mm plate / Detector: IMAGE PLATE / Date: Apr 1, 2000 / Details: BENT MIRROR |
| Radiation | Monochromator: TRIANGULAR SI / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.84 Å / Relative weight: 1 |
| Reflection | Resolution: 0.835→10 Å / Num. obs: 264548 / % possible obs: 93.8 % / Observed criterion σ(I): 0 / Redundancy: 3.9 % / Biso Wilson estimate: 5.9 Å2 / Rmerge(I) obs: 0.065 / Net I/σ(I): 7.7 |
| Reflection shell | Resolution: 0.835→0.85 Å / Redundancy: 1.7 % / Rmerge(I) obs: 0.379 / Mean I/σ(I) obs: 1.8 / % possible all: 77.8 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1E1A Resolution: 0.85→10 Å / Num. parameters: 29658 / Num. restraintsaints: 30675 / Cross valid method: FREE R / σ(F): 0 / Stereochemistry target values: ENGH AND HUBER Details: ANISOTROPIC REFINEMENT REDUCED FREE R (NO CUTOFF) BY 0.04
| |||||||||||||||||||||||||||||||||
| Refine analyze | Num. disordered residues: 63 / Occupancy sum hydrogen: 2047.3 / Occupancy sum non hydrogen: 2921.39 | |||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 0.85→10 Å
| |||||||||||||||||||||||||||||||||
| Refine LS restraints |
|
