 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1exi: CRYSTAL STRUCTURE OF TRANSCRIPTION ACTIVATOR BMRR, FROM B. SUBTIL... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1exi | ||||||
|---|---|---|---|---|---|---|---|
| Title | CRYSTAL STRUCTURE OF TRANSCRIPTION ACTIVATOR BMRR, FROM B. SUBTILIS, BOUND TO 21 BASE PAIR BMR OPERATOR AND TPSB | ||||||
 Components Components |
| ||||||
 Keywords Keywords | Transcription/DNA / Protein-DNA complex / MerR-family transcription activator / multidrug-binding protein / Transcription-DNA COMPLEX | ||||||
| Function / homology |  Function and homology information Function and homology informationDNA-binding transcription factor activity / regulation of DNA-templated transcription / DNA binding Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / Resolution: 3.12 Å X-RAY DIFFRACTION / Resolution: 3.12 Å | ||||||
 Authors Authors | Zheleznova-Heldwein, E.E. / Brennan, R.G. | ||||||
 Citation Citation | Journal: Nature / Year: 2001 Title: Crystal structure of the transcription activator BmrR bound to DNA and a drug. Authors: Heldwein, E.E. / Brennan, R.G. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1exi.cif.gz | 81.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1exi.ent.gz | 58.3 KB | Display | PDB format |
| PDBx/mmJSON format | 1exi.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ex/1exiftp://data.pdbj.org/pub/pdb/validation_reports/ex/1exi | HTTPS FTP |
|---|
-Related structure data











-Links
 PDBj
PDBj







































- Assembly
Assembly
| Deposited unit | 
| ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||||
| Unit cell |
| ||||||||||
| Components on special symmetry positions |
| ||||||||||
| Details | The biological assembly is a dimer constructed from chain A by generating a two-fold symmetry partner. / The biological assembly is a DNA duplex constructed from chain B by generating a two-fold symmetry partner. |
-Components
| #1: DNA chain | Mass: 6440.148 Da / Num. of mol.: 1 / Source method: obtained synthetically | ||
|---|---|---|---|
| #2: Protein | Mass: 32621.281 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) | ||
| #3: Chemical | ChemComp-ZN /   Mass: 65.409 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Zn Mass: 65.409 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Zn | ||
| #4: Chemical | 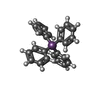  Mass: 430.176 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C24H20Sb Mass: 430.176 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C24H20Sb#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 3 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 3 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 5.61 Å3/Da / Density % sol: 78.09 % | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, hanging drop Details: 1 M imidazole, VAPOR DIFFUSION, HANGING DROP, temperature 298.0K | ||||||||||||||||||||
| Components of the solutions |
| ||||||||||||||||||||
| Crystal grow | *PLUS pH: 6.5 | ||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 298 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RU300 / Wavelength: 1.5418 |
| Detector | Type: RIGAKU RAXIS / Detector: IMAGE PLATE / Date: Feb 16, 2000 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 3.12→60.4 Å / Num. obs: 44719 / % possible obs: 86 % / Observed criterion σ(F): 1 / Observed criterion σ(I): 0.5 / Redundancy: 3.2 % / Rmerge(I) obs: 0.12 / Net I/σ(I): 3.7 |
| Reflection shell | Resolution: 3.12→3.6 Å / Redundancy: 2 % / Rmerge(I) obs: 0.278 / % possible all: 78 |
- Processing
Processing
| Software |
| ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 3.12→16 Å / Cross valid method: throughtout / σ(F): 1 / Stereochemistry target values: Engh & Huber
| ||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 3.12→16 Å
| ||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||
| Software | *PLUS Name: TNT / Classification: refinement | ||||||||||||||||||||
| Refinement | *PLUS σ(F): 1 / % reflection Rfree: 4.6 % | ||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||
| Refine LS restraints | *PLUS Type: t_angle_deg / Dev ideal: 1.5 |
