 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1cw8: SOLUTION STRUCTURE OF THE ANALOGUE RETRO-INVERSO (mA-R)REGRIGGC I... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1cw8 | ||||||
|---|---|---|---|---|---|---|---|
| Title | SOLUTION STRUCTURE OF THE ANALOGUE RETRO-INVERSO (mA-R)REGRIGGC IN CONTACT WITH THE MONOCLONAL ANTIBODY MAB 4X11, NMR, 6 STRUCTURES | ||||||
 Components Components | HISTONE H3 | ||||||
 Keywords Keywords | DNA BINDING PROTEIN / PSEUDOMIMETIC / SYNTHETIC PEPTIDE / RETRO-INVERSO ANALOGUE / TR-NOE / ANTIGEN- ANTIBODY COMPLEX | ||||||
| Function / homology | METHYLMALONIC ACID Function and homology information Function and homology information | ||||||
| Method | SOLUTION NMR / Energy minimisation Molecular dynamics (Simulated Annealing) | ||||||
 Authors Authors | Phan Chan Du, A. / Petit, M.C. / Guichard, G. / Briand, J.P. / Muller, S. / Cung, T. | ||||||
 Citation Citation | Journal: Biochemistry / Year: 2001 Title: Structure of antibody-bound peptides and retro-inverso analogues. A transferred nuclear Overhauser effect spectroscopy and molecular dynamics approach. Authors: Phan-Chan-Du, A. / Petit, M.C. / Guichard, G. / Briand, J.P. / Muller, S. / Cung, M.T. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1cw8.cif.gz | 21.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1cw8.ent.gz | 14.2 KB | Display | PDB format |
| PDBx/mmJSON format | 1cw8.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 1cw8_validation.pdf.gz | 352.9 KB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 1cw8_full_validation.pdf.gz | 362.3 KB | Display | |
| Data in XML | 1cw8_validation.xml.gz | 2.2 KB | Display | |
| Data in CIF | 1cw8_validation.cif.gz | 2.5 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/cw/1cw8ftp://data.pdbj.org/pub/pdb/validation_reports/cw/1cw8 | HTTPS FTP |
-Related structure data


-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| |||||||||
| NMR ensembles |
|
-Components
| #1: Protein/peptide | Mass: 846.980 Da / Num. of mol.: 1 / Fragment: C-TERMINAL DOMAIN 130-135 / Source method: obtained synthetically Details: The peptide was chemically synthesized. The sequence CGG was added as a linker to the dextran matrix in Biacore experiments via the Cys thiol group. NH2-CO was added at the C-terminal ...Details: The peptide was chemically synthesized. The sequence CGG was added as a linker to the dextran matrix in Biacore experiments via the Cys thiol group. NH2-CO was added at the C-terminal extremity of the retro-inverso peptide in order to mimic the N-terminal of the parent peptide. All non glycine residues present a D-configuration except CYS. Two RI(mA) diastereisomers were obtained and couldn't be separated. |
|---|---|
| #2: Chemical | ChemComp-DXX / 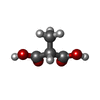  Mass: 118.088 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H6O4 Mass: 118.088 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H6O4 |
| Has protein modification | Y |
-Experimental details
-Experiment
| Experiment | Method: SOLUTION NMR |
|---|---|
| NMR experiment | Type: COSY, TOCSY, NOESY |
| NMR details | Text: The peptide/mAb molar ratio was adjusted to 50/1(i.e. 5 mM of peptide and 0.1 mM of mAb) |
- Sample preparation
Sample preparation
| Details | Contents: 5 mM peptide, 0.1 mM mAb; 100 mM phosphate buffer containning 0.02% sodium azide Solvent system: 95% H2O/5% D2O |
|---|---|
| Sample conditions | Ionic strength: 0.1M phosphate / pH: 7 / Pressure: 1 atm / Temperature: 277 K |
| Crystal grow | *PLUS Method: other / Details: NMR |
-NMR measurement
| NMR spectrometer | Type: Bruker DRX / Manufacturer: Bruker / Model: DRX / Field strength: 400 MHz |
|---|
- Processing
Processing
| NMR software |
| ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method: Energy minimisation Molecular dynamics (Simulated Annealing) Software ordinal: 1 Details: The structures are based on a set of 35 to 60 backbone-backbone, backbone-side chain and side chain-side chain distance restraints. The phi angle for the non glycine D-residues was ...Details: The structures are based on a set of 35 to 60 backbone-backbone, backbone-side chain and side chain-side chain distance restraints. The phi angle for the non glycine D-residues was constrained between 0 and 175. A distance dependent dielectric constant equal to 4r was applied. The net electric charges were decreased, while those of the N and C terminal charged groups were neglected. | ||||||||||||||||||||
| NMR representative | Selection criteria: lowest energy | ||||||||||||||||||||
| NMR ensemble | Conformer selection criteria: structures with the lowest energy Conformers calculated total number: 50 / Conformers submitted total number: 6 |
