 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1ba3 | ||||||
|---|---|---|---|---|---|---|---|
| Title | FIREFLY LUCIFERASE IN COMPLEX WITH BROMOFORM | ||||||
 Components Components | LUCIFERASE | ||||||
 Keywords Keywords | OXIDOREDUCTASE / MONOOXYGENASE / PHOTOPROTEIN / LUMINESCENCE | ||||||
| Function / homology |  Function and homology information Function and homology informationPhotinus-luciferin 4-monooxygenase (ATP-hydrolyzing) activity / firefly luciferase / CoA-ligase activity / bioluminescence / peroxisome / protein-folding chaperone binding / ATP binding / metal ion binding Similarity search - Function | ||||||
| Biological species |  Photinus pyralis (common eastern firefly) Photinus pyralis (common eastern firefly) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.2 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.2 Å | ||||||
 Authors Authors | Franks, N.P. / Jenkins, A. / Conti, E. / Lieb, W.R. / Brick, P. | ||||||
 Citation Citation | Journal: Biophys.J. / Year: 1998 Title: Structural basis for the inhibition of firefly luciferase by a general anesthetic. Authors: Franks, N.P. / Jenkins, A. / Conti, E. / Lieb, W.R. / Brick, P. #1: Journal: Embo J. / Year: 1997Title: Structural Basis for the Activation of Phenylalanine in the Non-Ribosomal Biosynthesis of Gramicidin S Authors: Conti, E. / Stachelhaus, T. / Marahiel, M.A. / Brick, P. #2: Journal: Structure / Year: 1996Title: Crystal Structure of Firefly Luciferase Throws Light on a Superfamily of Adenylate-Forming Enzymes Authors: Conti, E. / Franks, N.P. / Brick, P. #3: Journal: Acta Crystallogr.,Sect.D / Year: 1996Title: Crystallization and Preliminary Diffraction Studies of Firefly Luciferase from Photinus Pyralis Authors: Conti, E. / Lloyd, L.F. / Akins, J. / Franks, N.P. / Brick, P. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1ba3.cif.gz | 123.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1ba3.ent.gz | 94.8 KB | Display | PDB format |
| PDBx/mmJSON format | 1ba3.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 1ba3_validation.pdf.gz | 423.8 KB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 1ba3_full_validation.pdf.gz | 428 KB | Display | |
| Data in XML | 1ba3_validation.xml.gz | 24.1 KB | Display | |
| Data in CIF | 1ba3_validation.cif.gz | 35.4 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ba/1ba3ftp://data.pdbj.org/pub/pdb/validation_reports/ba/1ba3 | HTTPS FTP |
-Related structure data
| Related structure data |  1lciS S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 60818.953 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Photinus pyralis (common eastern firefly)Production host:  | ||
|---|---|---|---|
| #2: Chemical | 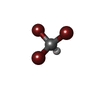  Mass: 252.731 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: CHBr3 Mass: 252.731 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: CHBr3#3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 353 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 353 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.8 Å3/Da / Density % sol: 56 % | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 283 K / Method: microbatch under oil / pH: 7.8 Details: 2 MICROLITRE OF LUCIFERASE (20 MG/ML) IN 0.2M AMMONIUM SULFATE, 0.001M EDTA, 0.001M DTT, 10% GLYCEROL, 25% ETHYLENE GLYCOL, 0.025M TRIS-HCL PH7.8 + 2 MICROLITRE 0.5M LITHIUM SULFATE, 26% PEG ...Details: 2 MICROLITRE OF LUCIFERASE (20 MG/ML) IN 0.2M AMMONIUM SULFATE, 0.001M EDTA, 0.001M DTT, 10% GLYCEROL, 25% ETHYLENE GLYCOL, 0.025M TRIS-HCL PH7.8 + 2 MICROLITRE 0.5M LITHIUM SULFATE, 26% PEG 8000, 0.1M TRIS-HCL PH7.8 AT 10 DEGREES CELSIUS IN MICROBATCH UNDER OIL., microbatch under oil, temperature 283K | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal | *PLUS | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 277 K / Method: vapor diffusionDetails: Conti, E., (1996) Acta Crystallogr.,Sect.D, 52, 876. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SRS  / Beamline: PX9.5 / Wavelength: 0.87 / Beamline: PX9.5 / Wavelength: 0.87 |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Jan 19, 1997 / Details: MIRRORS |
| Radiation | Monochromator: Y / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.87 Å / Relative weight: 1 |
| Reflection | Resolution: 2.2→18.4 Å / Num. obs: 35038 / % possible obs: 98.2 % / Redundancy: 3.13 % / Rmerge(I) obs: 0.071 / Rsym value: 0.071 / Net I/σ(I): 6.9 |
| Reflection shell | Resolution: 2.2→2.35 Å / Redundancy: 3.1 % / Rmerge(I) obs: 0.235 / Mean I/σ(I) obs: 3.1 / Rsym value: 0.235 / % possible all: 97.9 |
| Reflection | *PLUS Num. measured all: 109681 |
| Reflection shell | *PLUS % possible obs: 97.9 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 1LCI Resolution: 2.2→18.4 Å / Rfactor Rfree error: 0.006 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0 / Details: BULK SOLVENT CORRECTION APPLIED
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 29.9 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati d res low obs: 18.4 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.2→18.4 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.2→2.3 Å / Rfactor Rfree error: 0.02 / Total num. of bins used: 8
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Version: 3.851 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Rfactor Rfree: 0.3 |
