 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1ake: STRUCTURE OF THE COMPLEX BETWEEN ADENYLATE KINASE FROM ESCHERICHI... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1ake | ||||||
|---|---|---|---|---|---|---|---|
| Title | STRUCTURE OF THE COMPLEX BETWEEN ADENYLATE KINASE FROM ESCHERICHIA COLI AND THE INHIBITOR AP5A REFINED AT 1.9 ANGSTROMS RESOLUTION: A MODEL FOR A CATALYTIC TRANSITION STATE | ||||||
 Components Components | ADENYLATE KINASE | ||||||
 Keywords Keywords | TRANSFERASE(PHOSPHOTRANSFERASE) | ||||||
| Function / homology |  Function and homology information Function and homology informationpurine ribonucleotide interconversion / adenine metabolic process / nucleoside monophosphate metabolic process / ADP biosynthetic process / nucleoside diphosphate metabolic process / adenylate kinase / AMP kinase activity / AMP salvage / nucleoside diphosphate kinase activity / AMP binding ...purine ribonucleotide interconversion / adenine metabolic process / nucleoside monophosphate metabolic process / ADP biosynthetic process / nucleoside diphosphate metabolic process / adenylate kinase / AMP kinase activity / AMP salvage / nucleoside diphosphate kinase activity / AMP binding / magnesium ion binding / ATP binding / cytosol / cytoplasm Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / Resolution: 2 Å X-RAY DIFFRACTION / Resolution: 2 Å | ||||||
 Authors Authors | Mueller, C.W. / Schulz, G.E. | ||||||
 Citation Citation | Journal: J.Mol.Biol. / Year: 1992 Title: Structure of the complex between adenylate kinase from Escherichia coli and the inhibitor Ap5A refined at 1.9 A resolution. A model for a catalytic transition state. Authors: Muller, C.W. / Schulz, G.E. #1: Journal: J.Mol.Biol. / Year: 1990Title: Induced-Fit Movements in Adenylate Kinases Authors: Schulz, G.E. / Mueller, C.W. / Diederichs, K. #2: Journal: J.Mol.Biol. / Year: 1988Title: Structure of the Complex of Adenylate Kinase from Escherichia Coli with the Inhibitor P1, P5-Bis (Adenosine-5'-) Pentaphosphate Authors: Mueller, C.W. / Schulz, G.E. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1ake.cif.gz | 105.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1ake.ent.gz | 82.1 KB | Display | PDB format |
| PDBx/mmJSON format | 1ake.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ak/1akeftp://data.pdbj.org/pub/pdb/validation_reports/ak/1ake | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|








-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
| ||||||||
| Atom site foot note | 1: CIS PROLINE - PRO A 87 / 2: CIS PROLINE - PRO B 87 | ||||||||
| Noncrystallographic symmetry (NCS) | NCS oper: (Code: given Matrix: (-0.995341, 0.068333, 0.068023), Vector: Details | THE TRANSFORMATION PRESENTED ON THE *MTRIX* RECORDS WILL YIELD APPROXIMATE COORDINATES FOR CHAIN *A* WHEN APPLIED TO CHAIN *B*. | |
-Components
| #1: Protein | Mass: 23620.029 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) #2: Chemical | 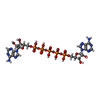  Mass: 916.367 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C20H29N10O22P5 Mass: 916.367 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C20H29N10O22P5#3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 378 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 378 / Source method: isolated from a natural source / Formula: H2OHas protein modification | N | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.63 Å3/Da / Density % sol: 53.17 % | ||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | *PLUS Temperature: 20 ℃ / pH: 6.7 / Method: vapor diffusion, hanging drop / Details: referred to J.Mol.Biol. 202.909-912 1988 | ||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Radiation | Scattering type: x-ray |
|---|---|
| Radiation wavelength | Relative weight: 1 |
| Reflection | *PLUS Highest resolution: 1.85 Å / Lowest resolution: 9999 Å / Num. obs: 40700 / % possible obs: 94.1 % / Num. measured all: 156159 / Rmerge(I) obs: 0.147 |
| Reflection shell | *PLUS Highest resolution: 1.85 Å / Lowest resolution: 1.98 Å / % possible obs: 92.8 % / Num. unique obs: 7170 / Rmerge(I) obs: 0.805 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 2→10 Å / Rfactor Rwork: 0.196 / Rfactor obs: 0.196 / σ(F): 0 Details: IN COMPLEX-I ARG 167 AND PHOSPHATE-4 OF AP5 ADOPT TWO CONFORMATIONS. BOTH CONFORMATIONS WERE REFINED ALTERNATINGLY. NOTE THAT THE DISTANCE PD - O3D FOR CONFORMATION A OF AP5 A 215 IS 2.13 ...Details: IN COMPLEX-I ARG 167 AND PHOSPHATE-4 OF AP5 ADOPT TWO CONFORMATIONS. BOTH CONFORMATIONS WERE REFINED ALTERNATINGLY. NOTE THAT THE DISTANCE PD - O3D FOR CONFORMATION A OF AP5 A 215 IS 2.13 ANGSTROMS WHICH IS LARGER THAN EXPECTED. NOTE FURTHER THAT CONFORMATION A OF THIS ENTRY CORRESPONDS TO CONFORMATION A' OF THE PAPER CITED ON JRNL RECORDS ABOVE AND CONFORMATION B CORRESPONDS TO CONFORMATION B' OF THE PUBLICATION. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2→10 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 1.9 Å / Lowest resolution: 10 Å / σ(F): 0 / Rfactor all: 0.196 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS Type: x_angle_d / Dev ideal: 3.2 |
