 ムービー
ムービー コントローラー
コントローラー


+ データを開く
データを開く

- 基本情報
基本情報
| 登録情報 |  | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| タイトル | Structure of SARS-CoV-2 Spike Protein with Engineered x3 Disulfide (x3(D427C, V987C) and single Arg S1/S2 cleavage site), incubated in Low pH after 40-Day Storage in PBS, Closed Conformation | ||||||||||||
 マップデータ マップデータ | |||||||||||||
 試料 試料 |
| ||||||||||||
| 生物種 |   Severe acute respiratory syndrome coronavirus 2 (ウイルス) Severe acute respiratory syndrome coronavirus 2 (ウイルス) | ||||||||||||
| 手法 | 単粒子再構成法 / クライオ電子顕微鏡法 / 解像度: 3.5 Å | ||||||||||||
 データ登録者 データ登録者 | Qu K / Chen Q / Ciazynska KA / Liu B / Zhang X / Wang J / He Y / Guan J / He J / Liu T ...Qu K / Chen Q / Ciazynska KA / Liu B / Zhang X / Wang J / He Y / Guan J / He J / Liu T / Carter AP / Xiong X / Briggs JAG | ||||||||||||
| 資金援助 | European Union,  英国, 3件 英国, 3件
| ||||||||||||
 引用 引用 | ジャーナル: PLoS Pathog / 年: 2022 タイトル: Engineered disulfide reveals structural dynamics of locked SARS-CoV-2 spike. 著者: Kun Qu / Qiuluan Chen / Katarzyna A Ciazynska / Banghui Liu / Xixi Zhang / Jingjing Wang / Yujie He / Jiali Guan / Jun He / Tian Liu / Xiaofei Zhang / Andrew P Carter / Xiaoli Xiong / John A G Briggs /    要旨: The spike (S) protein of SARS-CoV-2 has been observed in three distinct pre-fusion conformations: locked, closed and open. Of these, the function of the locked conformation remains poorly understood. ...The spike (S) protein of SARS-CoV-2 has been observed in three distinct pre-fusion conformations: locked, closed and open. Of these, the function of the locked conformation remains poorly understood. Here we engineered a SARS-CoV-2 S protein construct "S-R/x3" to arrest SARS-CoV-2 spikes in the locked conformation by a disulfide bond. Using this construct we determined high-resolution structures confirming that the x3 disulfide bond has the ability to stabilize the otherwise transient locked conformations. Structural analyses reveal that wild-type SARS-CoV-2 spike can adopt two distinct locked-1 and locked-2 conformations. For the D614G spike, based on which all variants of concern were evolved, only the locked-2 conformation was observed. Analysis of the structures suggests that rigidified domain D in the locked conformations interacts with the hinge to domain C and thereby restrains RBD movement. Structural change in domain D correlates with spike conformational change. We propose that the locked-1 and locked-2 conformations of S are present in the acidic high-lipid cellular compartments during virus assembly and egress. In this model, release of the virion into the neutral pH extracellular space would favour transition to the closed or open conformations. The dynamics of this transition can be altered by mutations that modulate domain D structure, as is the case for the D614G mutation, leading to changes in viral fitness. The S-R/x3 construct provides a tool for the further structural and functional characterization of the locked conformations of S, as well as how sequence changes might alter S assembly and regulation of receptor binding domain dynamics. | ||||||||||||
| 履歴 |
|
- 構造の表示
構造の表示
| 添付画像 |
|---|

- ダウンロードとリンク
ダウンロードとリンク
-EMDBアーカイブ
| マップデータ | emd_33462.map.gz | 104.1 MB |  EMDBマップデータ形式 EMDBマップデータ形式 | |
|---|---|---|---|---|
| ヘッダ (付随情報) | emd-33462-v30.xmlemd-33462.xml | 19.1 KB 19.1 KB | 表示 表示 | EMDBヘッダ |
| FSC (解像度算出) | emd_33462_fsc.xml | 12.8 KB | 表示 | FSCデータファイル |
| 画像 |  emd_33462.png emd_33462.png | 50.6 KB | ||
| その他 | emd_33462_half_map_1.map.gzemd_33462_half_map_2.map.gz | 140.7 MB 140.9 MB | ||
| アーカイブディレクトリ |  http://ftp.pdbj.org/pub/emdb/structures/EMD-33462ftp://ftp.pdbj.org/pub/emdb/structures/EMD-33462 http://ftp.pdbj.org/pub/emdb/structures/EMD-33462ftp://ftp.pdbj.org/pub/emdb/structures/EMD-33462 | HTTPS FTP |
-検証レポート
| 文書・要旨 | emd_33462_validation.pdf.gz | 685.2 KB | 表示 | EMDB検証レポート |
|---|---|---|---|---|
| 文書・詳細版 | emd_33462_full_validation.pdf.gz | 684.7 KB | 表示 | |
| XML形式データ | emd_33462_validation.xml.gz | 20.3 KB | 表示 | |
| CIF形式データ | emd_33462_validation.cif.gz | 26.7 KB | 表示 | |
| アーカイブディレクトリ | https://ftp.pdbj.org/pub/emdb/validation_reports/EMD-33462ftp://ftp.pdbj.org/pub/emdb/validation_reports/EMD-33462 | HTTPS FTP |
-関連構造データ



















-リンク
| EMDBのページ | EMDB (EBI/PDBe) / EMDataResource |
|---|
-マップ
| ファイル | ダウンロード / ファイル: emd_33462.map.gz / 形式: CCP4 / 大きさ: 178 MB / タイプ: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 投影像・断面図 | 画像のコントロール
画像は Spider により作成 | ||||||||||||||||||||||||||||||||||||
| ボクセルのサイズ | X=Y=Z: 1.061 Å | ||||||||||||||||||||||||||||||||||||
| 密度 |
| ||||||||||||||||||||||||||||||||||||
| 対称性 | 空間群: 1 | ||||||||||||||||||||||||||||||||||||
| 詳細 | EMDB XML:
|
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)




















-添付データ
-ハーフマップ: #1
| ファイル | emd_33462_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
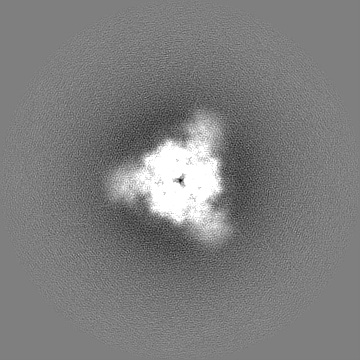
| 投影像・断面図 |
| ||||||||||||
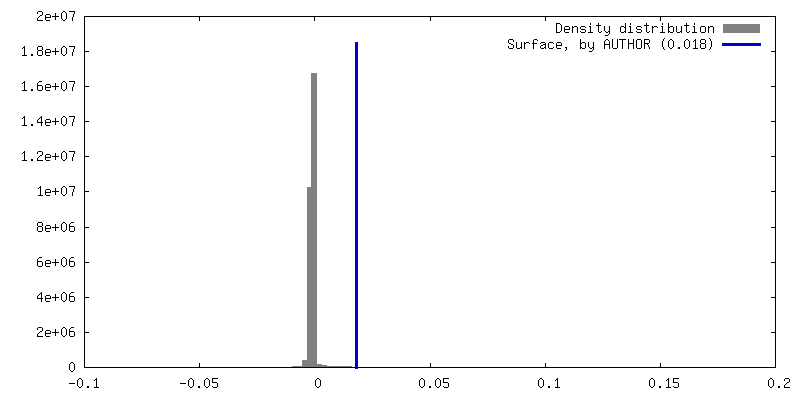
| 密度ヒストグラム |





-ハーフマップ: #2
| ファイル | emd_33462_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
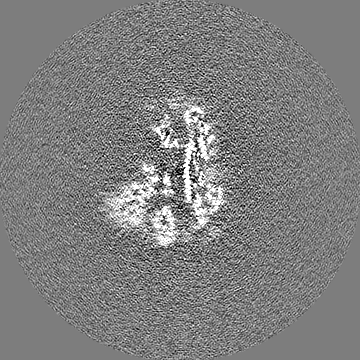
| 投影像・断面図 |
| ||||||||||||
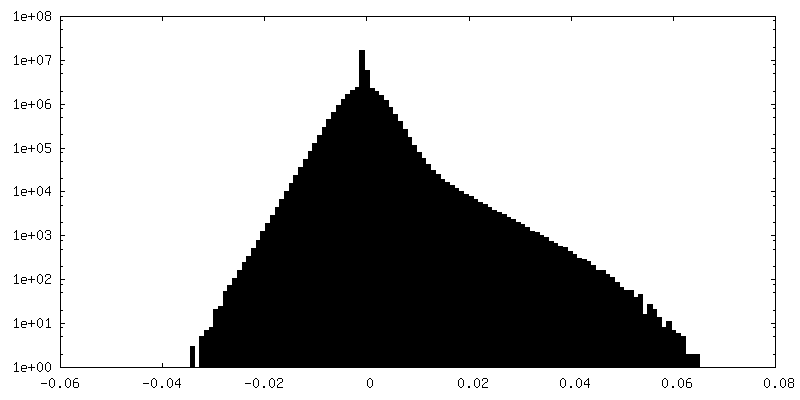
| 密度ヒストグラム |







- 試料の構成要素
試料の構成要素
-全体 : Severe acute respiratory syndrome coronavirus 2 Spike protein
| 全体 | 名称: Severe acute respiratory syndrome coronavirus 2 Spike protein |
|---|---|
| 要素 |
|
-超分子 #1: Severe acute respiratory syndrome coronavirus 2 Spike protein
| 超分子 | 名称: Severe acute respiratory syndrome coronavirus 2 Spike protein タイプ: complex / キメラ: Yes / ID: 1 / 親要素: 0 / 含まれる分子: all |
|---|---|
| 由来(天然) | 生物種: Severe acute respiratory syndrome coronavirus 2 (ウイルス) |
| 組換発現 | 生物種:  Homo sapiens (ヒト) Homo sapiens (ヒト) |
-分子 #1: Spike glycoprotein
| 分子 | 名称: Spike glycoprotein / タイプ: protein_or_peptide / ID: 1 / 光学異性体: LEVO |
|---|---|
| 由来(天然) | 生物種: Severe acute respiratory syndrome coronavirus 2 (ウイルス) |
| 組換発現 | 生物種: Homo sapiens (ヒト) |
| 配列 | 文字列: ETGTQCVNLT TRTQLPPAYT NSFTRGVYYP DKVFRSSVLH STQDLFLPFF SNVTWFHAIH VSGTNGTKRF DNPVLPFNDG VYFASTEKSN IIRGWIFGTT LDSKTQSLLI VNNATNVVIK VCEFQFCNDP FLGVYYHKNN KSWMESEFRV YSSANNCTFE YVSQPFLMDL ...文字列: ETGTQCVNLT TRTQLPPAYT NSFTRGVYYP DKVFRSSVLH STQDLFLPFF SNVTWFHAIH VSGTNGTKRF DNPVLPFNDG VYFASTEKSN IIRGWIFGTT LDSKTQSLLI VNNATNVVIK VCEFQFCNDP FLGVYYHKNN KSWMESEFRV YSSANNCTFE YVSQPFLMDL EGKQGNFKNL REFVFKNIDG YFKIYSKHTP INLVRDLPQG FSALEPLVDL PIGINITRFQ TLLALHRSYL TPGDSSSGWT AGAAAYYVGY LQPRTFLLKY NENGTITDAV DCALDPLSET KCTLKSFTVE KGIYQTSNFR VQPTESIVRF PNITNLCPFG EVFNATRFAS VYAWNRKRIS NCVADYSVLY NSASFSTFKC YGVSPTKLND LCFTNVYADS FVIRGDEVRQ IAPGQTGKIA DYNYKLPCDF TGCVIAWNSN NLDSKVGGNY NYLYRLFRKS NLKPFERDIS TEIYQAGSTP CNGVEGFNCY FPLQSYGFQP TNGVGYQPYR VVVLSFELLH APATVCGPKK STNLVKNKCV NFNFNGLTGT GVLTESNKKF LPFQQFGRDI ADTTDAVRDP QTLEILDITP CSFGGVSVIT PGTNTSNQVA VLYQDVNCTE VPVAIHADQL TPTWRVYSTG SNVFQTRAGC LIGAEHVNNS YECDIPIGAG ICASYQTQTN SRSVASQSII AYTMSLGAEN SVAYSNNSIA IPTNFTISVT TEILPVSMTK TSVDCTMYIC GDSTECSNLL LQYGSFCTQL NRALTGIAVE QDKNTQEVFA QVKQIYKTPP IKDFGGFNFS QILPDPSKPS KRSFIEDLLF NKVTLADAGF IKQYGDCLGD IAARDLICAQ KFNGLTVLPP LLTDEMIAQY TSALLAGTIT SGWTFGAGAA LQIPFAMQMA YRFNGIGVTQ NVLYENQKLI ANQFNSAIGK IQDSLSSTAS ALGKLQDVVN QNAQALNTLV KQLSSNFGAI SSVLNDILSR LDKCEAEVQI DRLITGRLQS LQTYVTQQLI RAAEIRASAN LAATKMSECV LGQSKRVDFC GKGYHLMSFP QSAPHGVVFL HVTYVPAQEK NFTTAPAICH DGKAHFPREG VFVSNGTHWF VTQRNFYEPQ IITTDNTFVS GNCDVVIGIV NNTVYDPLQP ELDSFKEELD KYFKNHTSPD VDLGDISGIN ASVVNIQKEI DRLNEVAKNL NESLIDLQEL GKYEQYIKGS GRENLYFQGG GGSGYIPEAP RDGQAYVRKD GEWVLLSTFL GHHHHHH |
-実験情報
-構造解析
| 手法 | クライオ電子顕微鏡法 |
|---|---|
 解析 解析 | 単粒子再構成法 |
| 試料の集合状態 | particle |
-試料調製
| 濃度 | 1.0 mg/mL |
|---|---|
| 緩衝液 | pH: 5 |
| グリッド | モデル: C-flat-2/2 / 支持フィルム - 材質: CARBON / 支持フィルム - トポロジー: HOLEY ARRAY / 前処理 - タイプ: GLOW DISCHARGE |
| 凍結 | 凍結剤: ETHANE / チャンバー内湿度: 100 % / チャンバー内温度: 298 K / 装置: FEI VITROBOT MARK IV |
- 電子顕微鏡法
電子顕微鏡法
| 顕微鏡 | FEI TITAN KRIOS |
|---|---|
| 特殊光学系 | エネルギーフィルター - 名称: GIF Quantum LS |
| 撮影 | フィルム・検出器のモデル: GATAN K3 BIOQUANTUM (6k x 4k) 撮影したグリッド数: 1 / 実像数: 1698 / 平均電子線量: 50.0 e/Å2 |
| 電子線 | 加速電圧: 300 kV / 電子線源:  FIELD EMISSION GUN FIELD EMISSION GUN |
| 電子光学系 | 照射モード: FLOOD BEAM / 撮影モード: BRIGHT FIELD / Cs: 2.7 mm / 最大 デフォーカス(公称値): 2.4 µm / 最小 デフォーカス(公称値): 1.0 µm / 倍率(公称値): 81000 |
| 試料ステージ | 試料ホルダーモデル: FEI TITAN KRIOS AUTOGRID HOLDER ホルダー冷却材: NITROGEN |
| 実験機器 |  モデル: Titan Krios / 画像提供: FEI Company |
+画像解析

-原子モデル構築 1
| 精密化 | 空間: REAL / プロトコル: RIGID BODY FIT / 当てはまり具合の基準: Correlation coefficient |
|---|
