Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-29502: Fast and versatile sequence- independent protein docking for nano... -
+ Open data
Open data

- Basic information
Basic information
| Entry |  | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Fast and versatile sequence- independent protein docking for nanomaterials design using RPXDock | |||||||||
 Map data Map data | Sharpened map | |||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords | octahedra / oligomer / de novo design / rosetta / cryoEM / interface / DE NOVO PROTEIN | |||||||||
| Biological species | synthetic construct (others) | |||||||||
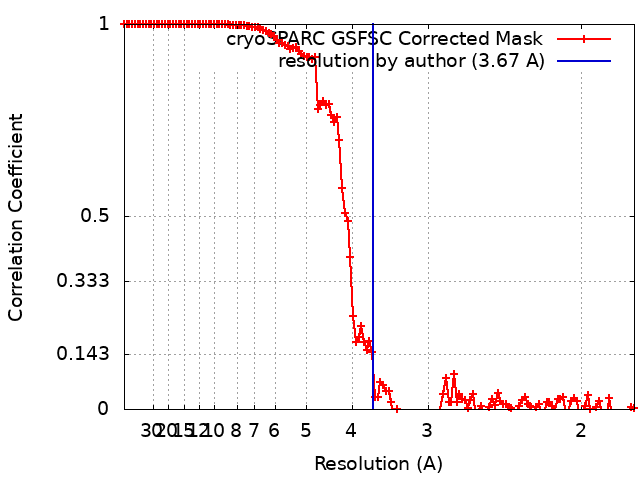
| Method | single particle reconstruction / cryo EM / Resolution: 3.67 Å | |||||||||
 Authors Authors | Skotheim R / Borst AJ / Baker D | |||||||||
| Funding support |  United States, 1 items United States, 1 items
| |||||||||
 Citation Citation | Journal: PLoS Comput Biol / Year: 2023 Title: Fast and versatile sequence-independent protein docking for nanomaterials design using RPXDock. Authors: William Sheffler / Erin C Yang / Quinton Dowling / Yang Hsia / Chelsea N Fries / Jenna Stanislaw / Mark D Langowski / Marisa Brandys / Zhe Li / Rebecca Skotheim / Andrew J Borst / Alena ...Authors: William Sheffler / Erin C Yang / Quinton Dowling / Yang Hsia / Chelsea N Fries / Jenna Stanislaw / Mark D Langowski / Marisa Brandys / Zhe Li / Rebecca Skotheim / Andrew J Borst / Alena Khmelinskaia / Neil P King / David Baker /  Abstract: Computationally designed multi-subunit assemblies have shown considerable promise for a variety of applications, including a new generation of potent vaccines. One of the major routes to such ...Computationally designed multi-subunit assemblies have shown considerable promise for a variety of applications, including a new generation of potent vaccines. One of the major routes to such materials is rigid body sequence-independent docking of cyclic oligomers into architectures with point group or lattice symmetries. Current methods for docking and designing such assemblies are tailored to specific classes of symmetry and are difficult to modify for novel applications. Here we describe RPXDock, a fast, flexible, and modular software package for sequence-independent rigid-body protein docking across a wide range of symmetric architectures that is easily customizable for further development. RPXDock uses an efficient hierarchical search and a residue-pair transform (RPX) scoring method to rapidly search through multidimensional docking space. We describe the structure of the software, provide practical guidelines for its use, and describe the available functionalities including a variety of score functions and filtering tools that can be used to guide and refine docking results towards desired configurations. | |||||||||
| History |
|
- Structure visualization
Structure visualization
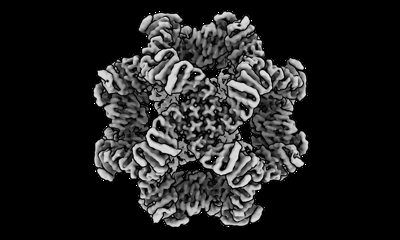
| Supplemental images |
|---|
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_29502.map.gz | 197.1 MB |  EMDB map data format EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-29502-v30.xmlemd-29502.xml | 23.2 KB 23.2 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_29502_fsc.xml | 12.6 KB | Display | FSC data file |
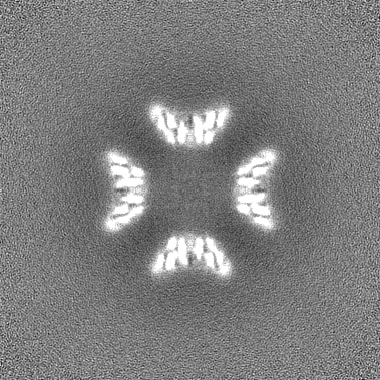
| Images |  emd_29502.png emd_29502.png | 69.7 KB | ||
| Filedesc metadata | emd-29502.cif.gz | 6.2 KB | ||
| Others | emd_29502_additional_1.map.gzemd_29502_additional_2.map.gzemd_29502_half_map_1.map.gzemd_29502_half_map_2.map.gz | 103.1 MB 178.7 MB 194 MB 194 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-29502ftp://ftp.pdbj.org/pub/emdb/structures/EMD-29502 http://ftp.pdbj.org/pub/emdb/structures/EMD-29502ftp://ftp.pdbj.org/pub/emdb/structures/EMD-29502 | HTTPS FTP |
-Related structure data

-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_29502.map.gz / Format: CCP4 / Size: 209.3 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Sharpened map | ||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 0.885 Å | ||||||||||||||||||||||||||||||||||||
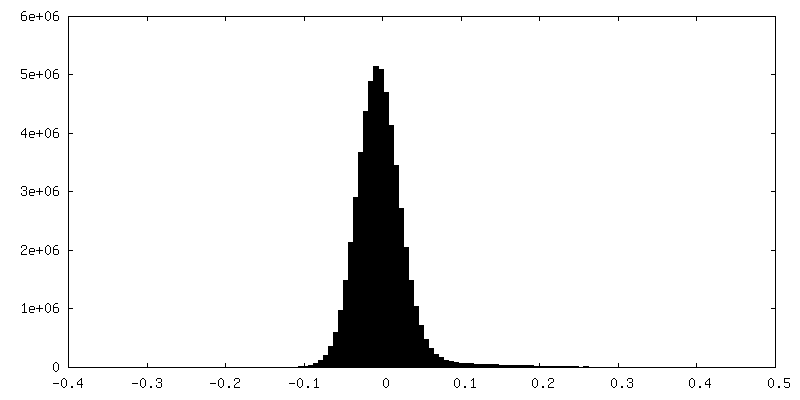
| Density |
| ||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
|
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















-Supplemental data
-Additional map: Unsharpened map
| File | emd_29502_additional_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Unsharpened map | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |






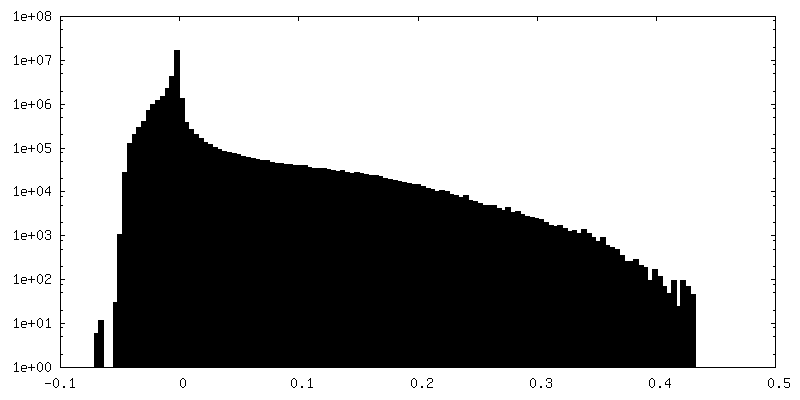
-Additional map: DeepEMhancer
| File | emd_29502_additional_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | DeepEMhancer | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |






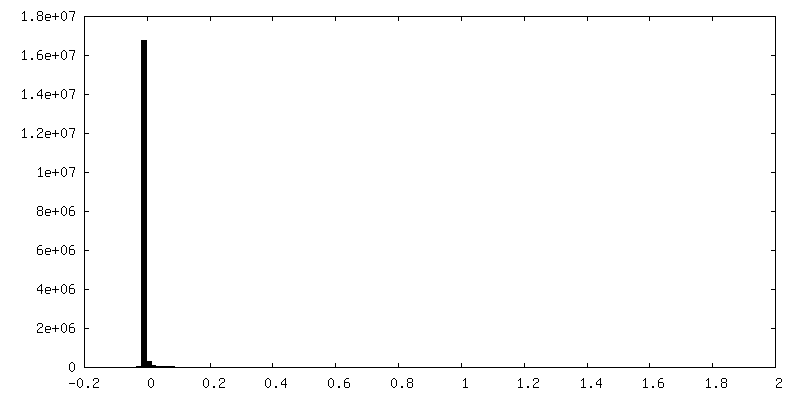
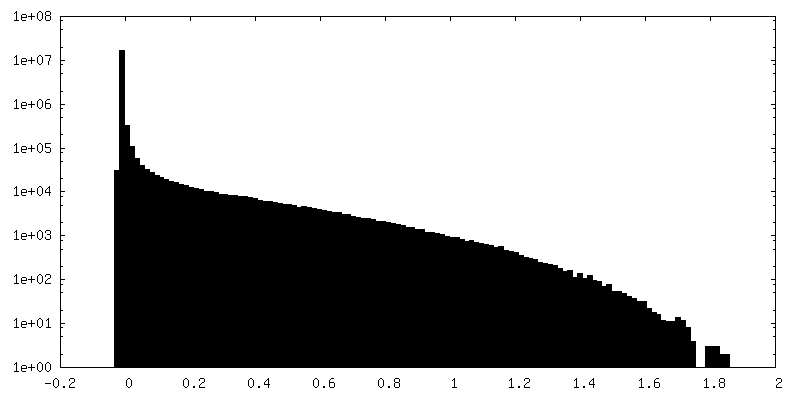
-Half map: Half map A
| File | emd_29502_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Half map A | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |



-Half map: Half map B
| File | emd_29502_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Half map B | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |



- Sample components
Sample components
-Entire : O43-rpxdock-EK1
| Entire | Name: O43-rpxdock-EK1 |
|---|---|
| Components |
|
-Supramolecule #1: O43-rpxdock-EK1
| Supramolecule | Name: O43-rpxdock-EK1 / type: complex / ID: 1 / Parent: 0 / Macromolecule list: all |
|---|---|
| Source (natural) | Organism: synthetic construct (others) |
-Macromolecule #1: O43-rpxdoc-EK1_A
| Macromolecule | Name: O43-rpxdoc-EK1_A / type: protein_or_peptide / ID: 1 / Number of copies: 24 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: synthetic construct (others) |
| Molecular weight | Theoretical: 26.624115 KDa |
| Recombinant expression | Organism:  |
| Sequence | String: EEAELAYLLG ELAYKLGEYR IAIRAYRIAL KRDPNNAEAW YNLGNAYTKQ GDYDEAIEYY LRALVLDPNN AEAATNLGQA YMNQGDKDR AKLMLLLALK LDPNNDSARV ILGVAKVGIE ELAKLASQAQ QEGDSEKQKA IELAAEAARV AQEVGDPELE K LALEAARR ...String: EEAELAYLLG ELAYKLGEYR IAIRAYRIAL KRDPNNAEAW YNLGNAYTKQ GDYDEAIEYY LRALVLDPNN AEAATNLGQA YMNQGDKDR AKLMLLLALK LDPNNDSARV ILGVAKVGIE ELAKLASQAQ QEGDSEKQKA IELAAEAARV AQEVGDPELE K LALEAARR GDSEKAKAIL LAAEAARVAK EVGDPELIKL ALEAARRGDS EKARAILEAA ERAREAKERG DPEQIKKARE LA KR |
-Macromolecule #2: O43-rpxdoc-EK1_B
| Macromolecule | Name: O43-rpxdoc-EK1_B / type: protein_or_peptide / ID: 2 / Number of copies: 24 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: synthetic construct (others) |
| Molecular weight | Theoretical: 15.268709 KDa |
| Recombinant expression | Organism: |
| Sequence | String: DVSALAYVML GLLLSLLNRL SLAAEAYKKA IELDPNDALA WLLLGSVLEK LKRLDEAAEA YKKAIELKPN DASAWKELGK VLEKLGRLD EAAEAYLIAI MLDPEDAEAA KELGKVLEKL GELEMAEEAY KLAIKLDPND |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 0.8 mg/mL |
|---|---|
| Buffer | pH: 7.5 |
| Grid | Model: Quantifoil R2/2 / Support film - Material: CARBON |
| Vitrification | Cryogen name: ETHANE |
- Electron microscopy
Electron microscopy
| Microscope | TFS GLACIOS |
|---|---|
| Image recording | Film or detector model: GATAN K3 (6k x 4k) / Average electron dose: 50.0 e/Å2 |
| Electron beam | Acceleration voltage: 200 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Nominal defocus max: 2.0 µm / Nominal defocus min: 1.0 µm |