 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-26682: Cryo-EM structure of the S. cerevisiae chromatin remodeler Yta7 h... -
+ Open data
Open data

- Basic information
Basic information
| Entry |  | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Cryo-EM structure of the S. cerevisiae chromatin remodeler Yta7 hexamer bound to ATPgS in state I | |||||||||
 Map data Map data | Primary map | |||||||||
 Sample Sample |
| |||||||||
| Biological species |  | |||||||||
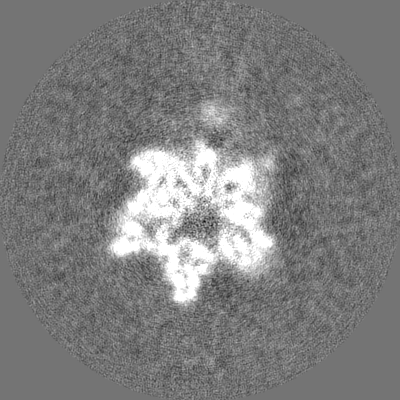
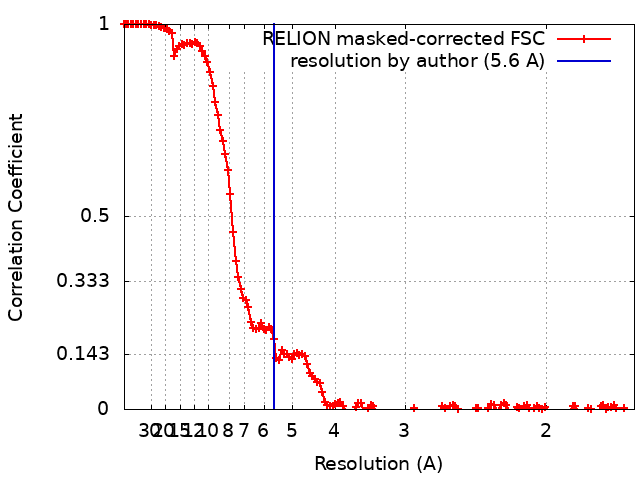
| Method | single particle reconstruction / cryo EM / Resolution: 5.6 Å | |||||||||
 Authors Authors | Wang F / Feng X / Li H | |||||||||
| Funding support |  United States, 1 items United States, 1 items
| |||||||||
 Citation Citation | Journal: J Biol Chem / Year: 2023 Title: The Saccharomyces cerevisiae Yta7 ATPase hexamer contains a unique bromodomain tier that functions in nucleosome disassembly. Authors: Feng Wang / Xiang Feng / Qing He / Hua Li / Huilin Li / Abstract: The Saccharomyces cerevisiae Yta7 is a chromatin remodeler harboring a histone-interacting bromodomain (BRD) and two AAA+ modules. It is not well understood how Yta7 recognizes the histone H3 tail to ...The Saccharomyces cerevisiae Yta7 is a chromatin remodeler harboring a histone-interacting bromodomain (BRD) and two AAA+ modules. It is not well understood how Yta7 recognizes the histone H3 tail to promote nucleosome disassembly for DNA replication or RNA transcription. By cryo-EM analysis, here we show that Yta7 assembles a three-tiered hexamer with a top BRD tier, a middle AAA1 tier, and a bottom AAA2 tier. Unexpectedly, the Yta7 BRD stabilizes a four-stranded β-helix, termed BRD-interacting motif (BIM), of the largely disordered N-terminal region. The BIM motif is unique to the baker's yeast, and we show both BRD and BIM contribute to nucleosome recognition. We found that Yta7 binds both acetylated and nonacetylated H3 peptides but with a higher affinity for the unmodified peptide. This property is consistent with the absence of key residues of canonical BRDs involved in acetylated peptide recognition and the role of Yta7 in general nucleosome remodeling. Interestingly, the BRD tier exists in a spiral and a flat-ring form on top of the Yta7 AAA+ hexamer. The spiral is likely in a nucleosome-searching mode because the bottom BRD blocks the entry to the AAA+ chamber. The flat ring may be in a nucleosome disassembly state because the entry is unblocked and the H3 peptide has entered the AAA+ chamber and is stabilized by the AAA1 pore loops 1 and 2. Indeed, we show that the BRD tier is a flat ring when bound to the nucleosome. Overall, our study sheds light on the nucleosome disassembly by Yta7. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Supplemental images |
|---|

- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_26682.map.gz | 206 MB |  EMDB map data format EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-26682-v30.xmlemd-26682.xml | 20.6 KB 20.6 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_26682_fsc.xml | 14.3 KB | Display | FSC data file |
| Images |  emd_26682.png emd_26682.png | 47.9 KB | ||
| Others | emd_26682_additional_1.map.gzemd_26682_half_map_1.map.gzemd_26682_half_map_2.map.gz | 192.1 MB 193.7 MB 193.5 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-26682ftp://ftp.pdbj.org/pub/emdb/structures/EMD-26682 http://ftp.pdbj.org/pub/emdb/structures/EMD-26682ftp://ftp.pdbj.org/pub/emdb/structures/EMD-26682 | HTTPS FTP |
-Validation report
| Summary document | emd_26682_validation.pdf.gz | 621.4 KB | Display | EMDB validaton report |
|---|---|---|---|---|
| Full document | emd_26682_full_validation.pdf.gz | 621 KB | Display | |
| Data in XML | emd_26682_validation.xml.gz | 21.6 KB | Display | |
| Data in CIF | emd_26682_validation.cif.gz | 28 KB | Display | |
| Arichive directory | https://ftp.pdbj.org/pub/emdb/validation_reports/EMD-26682ftp://ftp.pdbj.org/pub/emdb/validation_reports/EMD-26682 | HTTPS FTP |
-Related structure data







-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_26682.map.gz / Format: CCP4 / Size: 244.1 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Primary map | ||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 0.828 Å | ||||||||||||||||||||||||||||||||||||
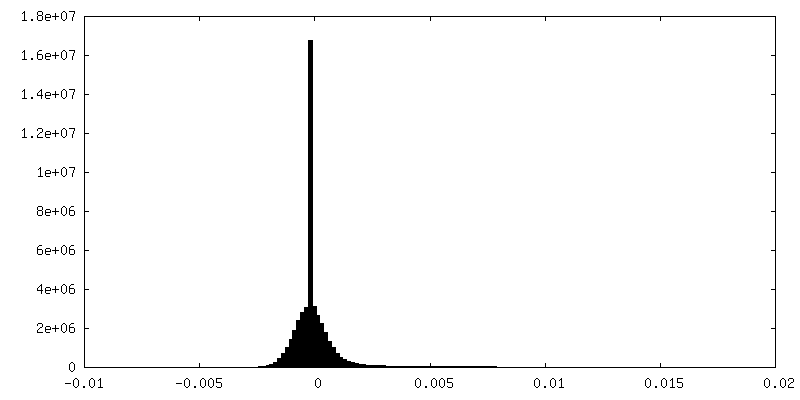
| Density |
| ||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
|
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















-Supplemental data
-Additional map: Raw map
| File | emd_26682_additional_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Raw map | ||||||||||||
| Projections & Slices |
| ||||||||||||
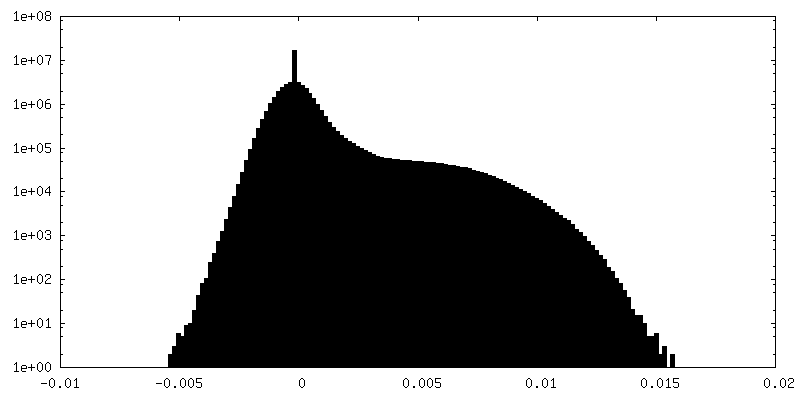
| Density Histograms |






-Half map: #1
| File | emd_26682_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
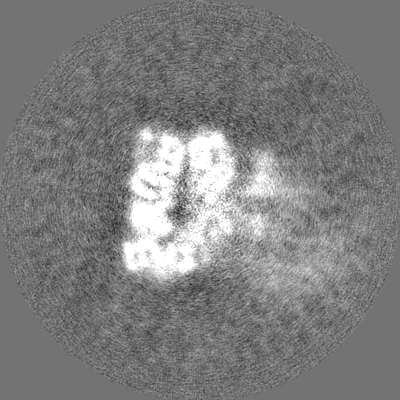
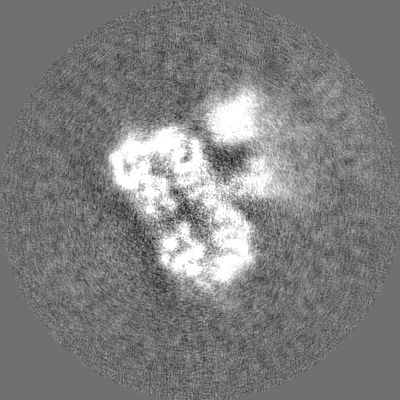
| Projections & Slices |
| ||||||||||||
| Density Histograms |




-Half map: #2
| File | emd_26682_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
| Density Histograms |



- Sample components
Sample components
-Entire : Complex of Yta7 and ATPgS (spiral BRD)
| Entire | Name: Complex of Yta7 and ATPgS (spiral BRD) |
|---|---|
| Components |
|
-Supramolecule #1: Complex of Yta7 and ATPgS (spiral BRD)
| Supramolecule | Name: Complex of Yta7 and ATPgS (spiral BRD) / type: complex / ID: 1 / Chimera: Yes / Parent: 0 / Macromolecule list: all |
|---|---|
| Source (natural) | Organism: |
| Molecular weight | Theoretical: 930 KDa |
-Macromolecule #1: Yta7
| Macromolecule | Name: Yta7 / type: protein_or_peptide / ID: 1 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: |
| Sequence | String: HHHHHHHHHH TSGSMDYKDH DGDYKDHDID YKDDDDKMAR NLRNRRGSDV EDASNAKVGY ETQIKDENGI IHTTTRSLRK INYAEIEKVF DFLEDDQVMD KDETPVDVTS DEHHNNNQKG DDEDDDVDLV SPHENARTNE ELTNERNLRK RKAHDPEEDD ESFHEEDVDD ...String: HHHHHHHHHH TSGSMDYKDH DGDYKDHDID YKDDDDKMAR NLRNRRGSDV EDASNAKVGY ETQIKDENGI IHTTTRSLRK INYAEIEKVF DFLEDDQVMD KDETPVDVTS DEHHNNNQKG DDEDDDVDLV SPHENARTNE ELTNERNLRK RKAHDPEEDD ESFHEEDVDD DEEEEEADEF EDEYLDEDSK DNNRRRRAAD RKFVVPDPDD DEEYDEDDEE GDRISHSASS KRLKRANSRR TRSSRHPETP PPVRRALRSR TRHSRTSNEE NDDENDNSRN EALTLADEIR ELQEDSPIRE KRFLRERTKP VNYKLPPPLT ASNAEEFIDK NNNALSFHNP SPARRGRGGW NASQNSGPTR RLFPTGGPFG GNDVTTIFGK NTNFYNQVPS AFSDNNNNKL ILDSDSSDDE ILPLGVTPKT KKENTQKKKK KKPEIADLDP LGVDMNVNFD DIGGLDNYID QLKEMVALPL LYPELYQNFN ITPPRGVLFH GPPGTGKTLM ARALAASCSS DERKITFFMR KGADILSKWV GEAERQLRLL FEEAKKHQPS IIFFDEIDGL APVRSSKQEQ IHASIVSTLL ALMDGMDNRG QVIVIGATNR PDAVDPALRR PGRFDREFYF PLPDVKARFK ILQIQTRKWS SPLSTNFIDK LAFLTKGYGG ADLRSLCTEA ALISIQRSFP QIYRSNDKLL VDPSKIKVKV SDFMLALKKI VPSSARSTGS SPQPLPELIK PLLADQLNNL KNKLDYMLNI KDTTFQRNTS LLQNFIDYEE YSGEEEEHDK YGGNEDTSSF RSYEFFESMA ESQICKPRLL INGPKGNGQQ YVGAAILNYL EEFNVQNLDL ASLVSESSRT IEAAVVQSFM EAKKRQPSVV FIPNLDIWIN TIPENVILVL SGLFRSLQSN EKILLLCLAE NLDISEVKNG ILSDFAFDKN IFQLHKPSKE NITRYFSNLI ELLKTKPSDI PMKKRRVKPL PELQKVTSNA APTNFDENGE PLSEKVVLRR KLKSFQHQDM RLKNVLKIKL SGLMDLFKNR YKRFRKPPID DAFLVHLFEP ETSNDPNWQP AYIKDENMIL EVSTGRKFFN MDLDIVEERL WNGYYSEPKQ FLKDIELIYR DANTIGDRER VIKASEMFAN AQMGIEEIST PDFIQECKAT RQRDLERQEL FLEDEEKRAA MELEAKEQSQ ENILQEPDLK DNKANEFGVA AGNQLQAQLQ TTINTASIVN NSEVPQPIDT NLYKKEIPAA IPSAVDKEKA VIPEDSGANE EYTTELIQAT CTSEITTDDD ERARKEPKEN EDSLQTQVTE ENFSKIDANT NNINHVKEIQ SVNKPNSLHE TVEKRERSPI PKEVVEPEQG KKSDKELILT PEQIKKVSAC LIEHCQNFTV SQLEDVHSSV AKIIWKSKSA WDKTGTVDEI IKFLSE |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 10 mg/mL | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Buffer | pH: 7.6 Component:
Details: Solution was made fresh and detergent was added to solve preference orientation issue. | ||||||||||||||||||||||||
| Grid | Model: Quantifoil R2/1 / Material: COPPER / Mesh: 300 / Support film - Material: GOLD / Support film - topology: CONTINUOUS / Pretreatment - Type: GLOW DISCHARGE / Pretreatment - Time: 30 sec. / Pretreatment - Atmosphere: AIR / Pretreatment - Pressure: 101.325 kPa | ||||||||||||||||||||||||
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 100 % / Chamber temperature: 281 K / Instrument: FEI VITROBOT MARK IV / Details: Blot 3S, blot forth 3. | ||||||||||||||||||||||||
| Details | The sample was a novel chromatin remodeler and a AAA+ ATPase. |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Temperature | Min: 193.0 K / Max: 193.0 K |
| Image recording | Film or detector model: GATAN K3 (6k x 4k) / Average electron dose: 65.0 e/Å2 Details: A total of 75 frames were recorded for each micrograph stack. |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | C2 aperture diameter: 70.0 µm / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2.7 mm / Nominal defocus max: 2.0 µm / Nominal defocus min: 1.0 µm / Nominal magnification: 105000 |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER / Cooling holder cryogen: NITROGEN |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
+Image processing

-Atomic model buiding 1
| Refinement | Space: REAL / Protocol: RIGID BODY FIT / Target criteria: Correlation coefficient |
|---|
