- EMDB-26674: Human mitochondrial AAA protein ATAD1 (with a catalytic dead muta... -
+
Open data
ID or keywords:
Loading...
-
Basic information
Entry
Database: EMDB / ID: EMD-26674
Title
Human mitochondrial AAA protein ATAD1 (with a catalytic dead mutation) in complex with a peptide substrate (closed conformation)
Map data
Human mitochondrial AAA protein ATAD1
Sample
Complex: ATAD1-substrate complex in closed conformation
Protein or peptide: Outer mitochondrial transmembrane helix translocase
Protein or peptide: Unknown peptide substrate
Ligand: ADENOSINE-5'-TRIPHOSPHATE
Ligand: MAGNESIUM ION
Keywords
AAA protein / mitochondria / tail-anchored protein / membrane protein / PROTEIN TRANSPORT
Function / homology
Function and homology information
extraction of mislocalized protein from mitochondrial outer membrane / Class I peroxisomal membrane protein import / membrane protein dislocase activity / Translocases; Catalysing the translocation of amino acids and peptides; Linked to the hydrolysis of a nucleoside triphosphate / peroxisomal membrane / negative regulation of synaptic transmission, glutamatergic / regulation of postsynaptic neurotransmitter receptor internalization / positive regulation of receptor internalization / learning / memory ...extraction of mislocalized protein from mitochondrial outer membrane / Class I peroxisomal membrane protein import / membrane protein dislocase activity / Translocases; Catalysing the translocation of amino acids and peptides; Linked to the hydrolysis of a nucleoside triphosphate / peroxisomal membrane / negative regulation of synaptic transmission, glutamatergic / regulation of postsynaptic neurotransmitter receptor internalization / positive regulation of receptor internalization / learning / memory / mitochondrial outer membrane / postsynaptic membrane / glutamatergic synapse / ATP hydrolysis activity / mitochondrion / ATP binding / membrane / cytosol Similarity search - Function
: / AAA ATPase, AAA+ lid domain / AAA+ lid domain / ATPase, AAA-type, conserved site / AAA-protein family signature. / ATPase family associated with various cellular activities (AAA) / ATPase, AAA-type, core / ATPases associated with a variety of cellular activities / AAA+ ATPase domain / P-loop containing nucleoside triphosphate hydrolase Similarity search - Domain/homology
National Institutes of Health/National Institute of General Medical Sciences (NIH/NIGMS)
R01GM032384
United States
National Institutes of Health/National Institute of General Medical Sciences (NIH/NIGMS)
S10OD021741
United States
National Institutes of Health/National Institute of General Medical Sciences (NIH/NIGMS)
S10OD020054
United States
Citation
Journal: Elife / Year: 2022 Title: Conserved structural elements specialize ATAD1 as a membrane protein extraction machine. Authors: Lan Wang / Hannah Toutkoushian / Vladislav Belyy / Claire Y Kokontis / Peter Walter / Abstract: The mitochondrial AAA (TPase ssociated with diverse cellular ctivities) protein ATAD1 (in humans; Msp1 in yeast) removes mislocalized membrane proteins, as well as stuck import substrates from the ...The mitochondrial AAA (TPase ssociated with diverse cellular ctivities) protein ATAD1 (in humans; Msp1 in yeast) removes mislocalized membrane proteins, as well as stuck import substrates from the mitochondrial outer membrane, facilitating their re-insertion into their cognate organelles and maintaining mitochondria's protein import capacity. In doing so, it helps to maintain proteostasis in mitochondria. How ATAD1 tackles the energetic challenge to extract hydrophobic membrane proteins from the lipid bilayer and what structural features adapt ATAD1 for its particular function has remained a mystery. Previously, we determined the structure of Msp1 in complex with a peptide substrate (Wang et al., 2020). The structure showed that Msp1's mechanism follows the general principle established for AAA proteins while adopting several structural features that specialize it for its function. Among these features in Msp1 was the utilization of multiple aromatic amino acids to firmly grip the substrate in the central pore. However, it was not clear whether the aromatic nature of these amino acids were required, or if they could be functionally replaced by aliphatic amino acids. In this work, we determined the cryo-EM structures of the human ATAD1 in complex with a peptide substrate at near atomic resolution. The structures show that phylogenetically conserved structural elements adapt ATAD1 for its function while generally adopting a conserved mechanism shared by many AAA proteins. We developed a microscopy-based assay reporting on protein mislocalization, with which we directly assessed ATAD1's activity in live cells and showed that both aromatic amino acids in pore-loop 1 are required for ATAD1's function and cannot be substituted by aliphatic amino acids. A short α-helix at the C-terminus strongly facilitates ATAD1's oligomerization, a structural feature that distinguishes ATAD1 from its closely related proteins.
Name: Outer mitochondrial transmembrane helix translocase / type: protein_or_peptide / ID: 1 / Number of copies: 6 / Enantiomer: LEVO EC number: Translocases; Catalysing the translocation of amino acids and peptides; Linked to the hydrolysis of a nucleoside triphosphate
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information
 Map data
Map data Sample
Sample Keywords
Keywords Function and homology information
Function and homology information Homo sapiens (human) /
Homo sapiens (human) / 
 Authors
Authors United States, 3 items
United States, 3 items  Citation
Citation Structure visualization
Structure visualization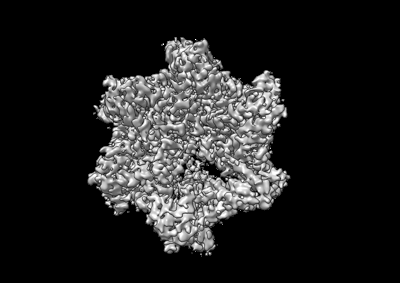
 Downloads & links
Downloads & links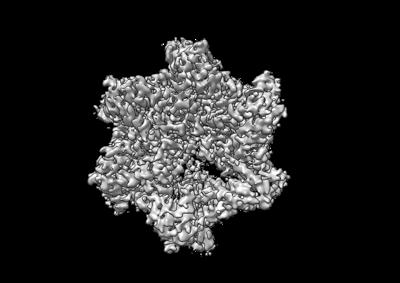 emd_26674.png
emd_26674.png http://ftp.pdbj.org/pub/emdb/structures/EMD-26674
http://ftp.pdbj.org/pub/emdb/structures/EMD-26674





 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)























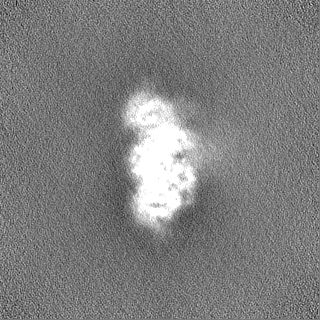
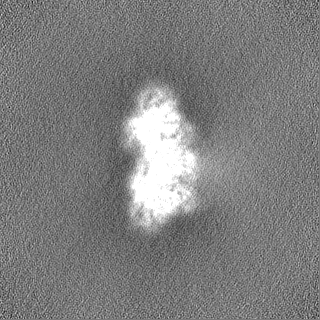
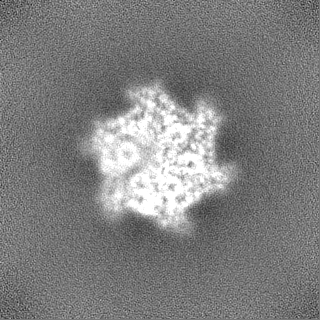
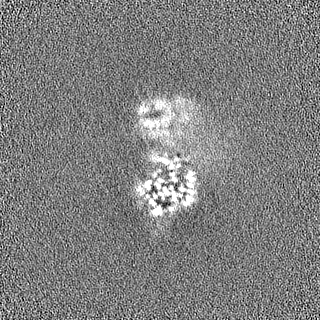
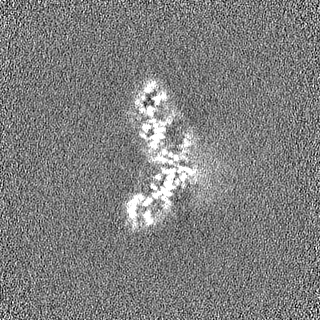
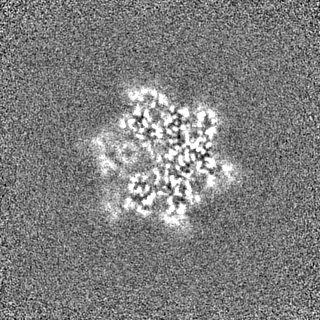
 Sample components
Sample components
 Processing
Processing Electron microscopy
Electron microscopy FIELD EMISSION GUN
FIELD EMISSION GUN
