 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-25468: MicroED structure of proteinase K from a 320 nm thick lamella mea... -
+ Open data
Open data

- Basic information
Basic information
| Entry |  | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | MicroED structure of proteinase K from a 320 nm thick lamella measured at 300 kV | |||||||||
 Map data Map data | 2Fo-Fc MicroED map from a 320 nm thick lamella measured at 300 kV | |||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords | Serine protease / HYDROLASE | |||||||||
| Function / homology |  Function and homology information Function and homology informationpeptidase K / serine-type endopeptidase activity / proteolysis / extracellular space / metal ion binding Similarity search - Function | |||||||||
| Biological species |  Parengyodontium album (fungus) Parengyodontium album (fungus) | |||||||||
| Method | electron crystallography / cryo EM | |||||||||
 Authors Authors | Martynowycz MW / Clabbers MTB | |||||||||
| Funding support |  United States, 2 items United States, 2 items
| |||||||||
 Citation Citation | Journal: Proc Natl Acad Sci U S A / Year: 2021 Title: Benchmarking the ideal sample thickness in cryo-EM. Authors: Michael W Martynowycz / Max T B Clabbers / Johan Unge / Johan Hattne / Tamir Gonen / Abstract: The relationship between sample thickness and quality of data obtained is investigated by microcrystal electron diffraction (MicroED). Several electron microscopy (EM) grids containing proteinase K ...The relationship between sample thickness and quality of data obtained is investigated by microcrystal electron diffraction (MicroED). Several electron microscopy (EM) grids containing proteinase K microcrystals of similar sizes from the same crystallization batch were prepared. Each grid was transferred into a focused ion beam and a scanning electron microscope in which the crystals were then systematically thinned into lamellae between 95- and 1,650-nm thick. MicroED data were collected at either 120-, 200-, or 300-kV accelerating voltages. Lamellae thicknesses were expressed in multiples of the corresponding inelastic mean free path to allow the results from different acceleration voltages to be compared. The quality of the data and subsequently determined structures were assessed using standard crystallographic measures. Structures were reliably determined with similar quality from crystalline lamellae up to twice the inelastic mean free path. Lower resolution diffraction was observed at three times the mean free path for all three accelerating voltages, but the data quality was insufficient to yield structures. Finally, no coherent diffraction was observed from lamellae thicker than four times the calculated inelastic mean free path. This study benchmarks the ideal specimen thickness with implications for all cryo-EM methods. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Supplemental images |
|---|

- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_25468.map.gz | 3.9 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-25468-v30.xmlemd-25468.xml | 14 KB 14 KB | Display Display | EMDB header |
| Images | 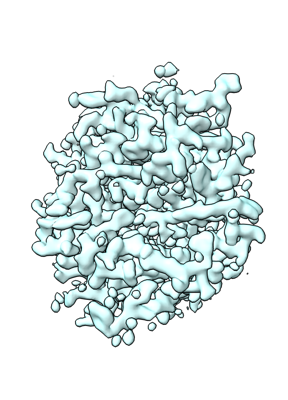 emd_25468.png emd_25468.png | 100.8 KB | ||
| Filedesc metadata | emd-25468.cif.gz | 5.6 KB | ||
| Filedesc structureFactors | emd_25468_sf.cif.gz | 508.9 KB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-25468ftp://ftp.pdbj.org/pub/emdb/structures/EMD-25468 http://ftp.pdbj.org/pub/emdb/structures/EMD-25468ftp://ftp.pdbj.org/pub/emdb/structures/EMD-25468 | HTTPS FTP |
-Validation report
| Summary document | emd_25468_validation.pdf.gz | 468.1 KB | Display | EMDB validaton report |
|---|---|---|---|---|
| Full document | emd_25468_full_validation.pdf.gz | 467.7 KB | Display | |
| Data in XML | emd_25468_validation.xml.gz | 4.5 KB | Display | |
| Data in CIF | emd_25468_validation.cif.gz | 5 KB | Display | |
| Arichive directory | https://ftp.pdbj.org/pub/emdb/validation_reports/EMD-25468ftp://ftp.pdbj.org/pub/emdb/validation_reports/EMD-25468 | HTTPS FTP |
-Related structure data
| Related structure data |  7swaMC  7svyC  7svzC  7sw0C  7sw1C  7sw2C  7sw3C  7sw4C  7sw5C  7sw6C  7sw7C  7sw8C  7sw9C  7swbC  7swcC M: atomic model generated by this map C: citing same article ( |
|---|---|
| Similar structure data |













-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| Related items in Molecule of the Month |


-Map
| File | Download / File: emd_25468.map.gz / Format: CCP4 / Size: 4.2 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | 2Fo-Fc MicroED map from a 320 nm thick lamella measured at 300 kV | ||||||||||||||||||||
| Voxel size | X: 0.50622 Å / Y: 0.50622 Å / Z: 0.48667 Å | ||||||||||||||||||||
| Density |
| ||||||||||||||||||||
| Symmetry | Space group: 96 | ||||||||||||||||||||
| Details | EMDB XML:
|
-Supplemental data
- Sample components
Sample components
-Entire : Proteinase K
| Entire | Name: Proteinase K |
|---|---|
| Components |
|
-Supramolecule #1: Proteinase K
| Supramolecule | Name: Proteinase K / type: complex / ID: 1 / Parent: 0 / Macromolecule list: #1 / Details: Serine protease |
|---|---|
| Source (natural) | Organism: Parengyodontium album (fungus) |
| Molecular weight | Theoretical: 28.9 KDa |
-Macromolecule #1: Proteinase K
| Macromolecule | Name: Proteinase K / type: protein_or_peptide / ID: 1 / Number of copies: 1 / Enantiomer: LEVO / EC number: peptidase K |
|---|---|
| Source (natural) | Organism: Parengyodontium album (fungus) |
| Molecular weight | Theoretical: 28.930783 KDa |
| Sequence | String: AAQTNAPWGL ARISSTSPGT STYYYDESAG QGSCVYVIDT GIEASHPEFE GRAQMVKTYY YSSRDGNGHG THCAGTVGSR TYGVAKKTQ LFGVKVLDDN GSGQYSTIIA GMDFVASDKN NRNCPKGVVA SLSLGGGYSS SVNSAAARLQ SSGVMVAVAA G NNNADARN ...String: AAQTNAPWGL ARISSTSPGT STYYYDESAG QGSCVYVIDT GIEASHPEFE GRAQMVKTYY YSSRDGNGHG THCAGTVGSR TYGVAKKTQ LFGVKVLDDN GSGQYSTIIA GMDFVASDKN NRNCPKGVVA SLSLGGGYSS SVNSAAARLQ SSGVMVAVAA G NNNADARN YSPASEPSVC TVGASDRYDR RSSFSNYGSV LDIFGPGTSI LSTWIGGSTR SISGTSMATP HVAGLAAYLM TL GKTTAAS ACRYIADTAN KGDLSNIPFG TVNLLAYNNY QA UniProtKB: Proteinase K |
-Macromolecule #2: water
| Macromolecule | Name: water / type: ligand / ID: 2 / Number of copies: 101 / Formula: HOH |
|---|---|
| Molecular weight | Theoretical: 18.015 Da |
| Chemical component information |  ChemComp-HOH: |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | electron crystallography |
| Aggregation state | 3D array |
-Sample preparation
| Concentration | 5 mg/mL |
|---|---|
| Buffer | pH: 7.5 |
| Grid | Model: Quantifoil R2/2 / Material: COPPER / Mesh: 200 / Support film - Material: CARBON / Support film - topology: HOLEY / Support film - Film thickness: 10 / Pretreatment - Type: GLOW DISCHARGE / Pretreatment - Time: 30 sec. |
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 95 % / Chamber temperature: 277 K / Instrument: LEICA PLUNGER |
| Details | Microcrystals |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Temperature | Min: 77.0 K / Max: 90.0 K |
| Image recording | Film or detector model: FEI CETA (4k x 4k) / Digitization - Dimensions - Width: 2048 pixel / Digitization - Dimensions - Height: 2048 pixel / Number grids imaged: 1 / Number real images: 1 / Number diffraction images: 120 / Average exposure time: 1.0 sec. / Average electron dose: 0.01 e/Å2 Details: 0.5 degrees per second, 1 second readout, 30 to -30 degrees. |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | C2 aperture diameter: 50.0 µm / Illumination mode: FLOOD BEAM / Imaging mode: DIFFRACTION / Cs: 2.7 mm / Camera length: 2460 mm |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER / Cooling holder cryogen: NITROGEN |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
-Image processing
| Details | Binned by 2. |
|---|---|
| Final reconstruction | Resolution method: DIFFRACTION PATTERN/LAYERLINES / Software - Name: PHENIX |
| Merging software list | Software - Name: AIMLESS |
| Crystallography statistics | Number intensities measured: 71235 / Number structure factors: 22907 / Fourier space coverage: 83 / R sym: 14 / R merge: 30 / Overall phase error: 25 / Overall phase residual: 0 / Phase error rejection criteria: None / High resolution: 2.1 Å / Shell - Shell ID: 1 / Shell - High resolution: 2.1 Å / Shell - Low resolution: 2.18 Å / Shell - Number structure factors: 2161 / Shell - Phase residual: 34 / Shell - Fourier space coverage: 84 / Shell - Multiplicity: 3 |
-Atomic model buiding 1
| Initial model | PDB ID: Chain - Chain ID: A / Chain - Residue range: 106-384 / Chain - Source name: PDB / Chain - Initial model type: experimental model |
|---|---|
| Refinement | Space: RECIPROCAL / Protocol: RIGID BODY FIT / Overall B value: 19 / Target criteria: Maximum likelihood |
| Output model | PDB-7swa: |

