National Institutes of Health/National Institute of General Medical Sciences (NIH/NIGMS)
R01GM080139
United States
Citation
Journal: Nat Methods / Year: 2021 Title: Deep learning-based mixed-dimensional Gaussian mixture model for characterizing variability in cryo-EM. Authors: Muyuan Chen / Steven J Ludtke / Abstract: Structural flexibility and/or dynamic interactions with other molecules is a critical aspect of protein function. Cryogenic electron microscopy (cryo-EM) provides direct visualization of individual ...Structural flexibility and/or dynamic interactions with other molecules is a critical aspect of protein function. Cryogenic electron microscopy (cryo-EM) provides direct visualization of individual macromolecules sampling different conformational and compositional states. While numerous methods are available for computational classification of discrete states, characterization of continuous conformational changes or large numbers of discrete state without human supervision remains challenging. Here we present e2gmm, a machine learning algorithm to determine a conformational landscape for proteins or complexes using a three-dimensional Gaussian mixture model mapped onto two-dimensional particle images in known orientations. Using a deep neural network architecture, e2gmm can automatically resolve the structural heterogeneity within the protein complex and map particles onto a small latent space describing conformational and compositional changes. This system presents a more intuitive and flexible representation than other manifold methods currently in use. We demonstrate this method on both simulated data and three biological systems to explore compositional and conformational changes at a range of scales. The software is distributed as part of EMAN2.
History
Deposition
May 27, 2021
-
Header (metadata) release
Aug 25, 2021
-
Map release
Aug 25, 2021
-
Update
Aug 25, 2021
-
Current status
Aug 25, 2021
Processing site: RCSB / Status: Released
-
Structure visualization
Movie
Surface view with section colored by density value
Organism: Severe acute respiratory syndrome coronavirus 2
-
Experimental details
-
Structure determination
Method
cryo EM
Processing
single particle reconstruction
Aggregation state
particle
-
Sample preparation
Buffer
pH: 7
Vitrification
Cryogen name: ETHANE
-
Electron microscopy
Microscope
FEI TITAN KRIOS
Image recording
Film or detector model: GATAN K2 SUMMIT (4k x 4k) / Average electron dose: 50.0 e/Å2
Electron beam
Acceleration voltage: 300 kV / Electron source: FIELD EMISSION GUN
Electron optics
Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD
Experimental equipment
Model: Titan Krios / Image courtesy: FEI Company
-
Image processing
Final reconstruction
Resolution.type: BY AUTHOR / Resolution: 5.0 Å / Resolution method: OTHER Details: 10000 particles are used to reconstruct each frame of the movie. The maps are filtered to 5A for display Number images used: 10000
Initial angle assignment
Type: MAXIMUM LIKELIHOOD
Final angle assignment
Type: MAXIMUM LIKELIHOOD / Software - Name: RELION
Final 3D classification
Software - Name: EMAN (ver. 2.91)
+
About Yorodumi
-
News
-
Feb 9, 2022. New format data for meta-information of EMDB entries
New format data for meta-information of EMDB entries
Version 3 of the EMDB header file is now the official format.
The previous official version 1.9 will be removed from the archive.
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Map data
Map data Sample
Sample Function and homology information
Function and homology information
 Severe acute respiratory syndrome coronavirus 2
Severe acute respiratory syndrome coronavirus 2 Authors
Authors United States, 1 items
United States, 1 items  Citation
Citation Structure visualization
Structure visualization
 Downloads & links
Downloads & links emd_24119.png
emd_24119.png http://ftp.pdbj.org/pub/emdb/structures/EMD-24119
http://ftp.pdbj.org/pub/emdb/structures/EMD-24119





















 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)










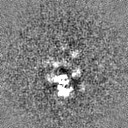

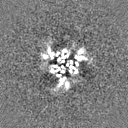









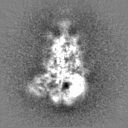
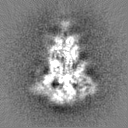
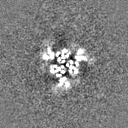

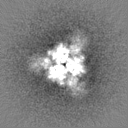
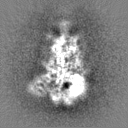
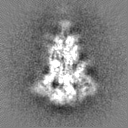
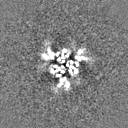
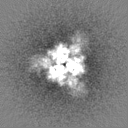
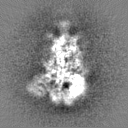
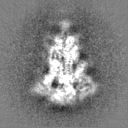
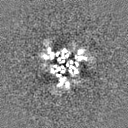


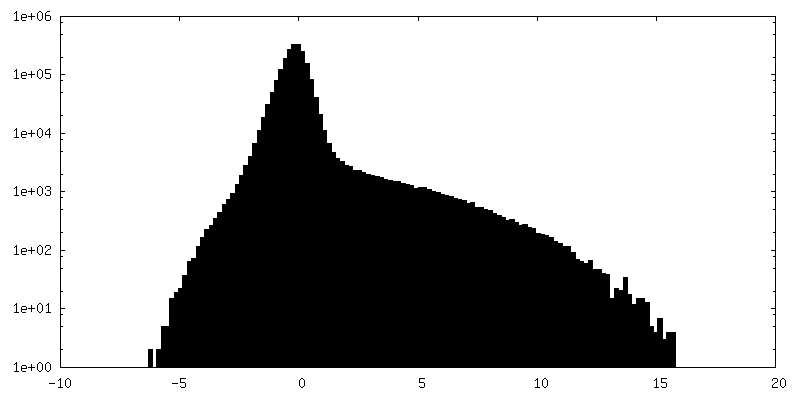






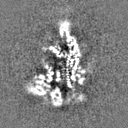
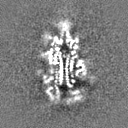
 Sample components
Sample components Processing
Processing Electron microscopy
Electron microscopy FIELD EMISSION GUN
FIELD EMISSION GUN
