Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
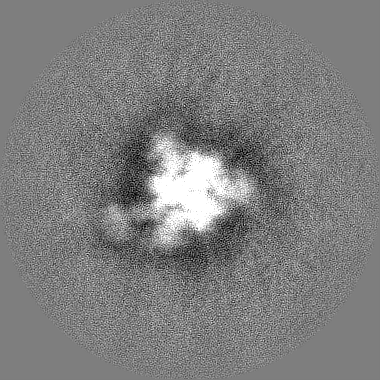
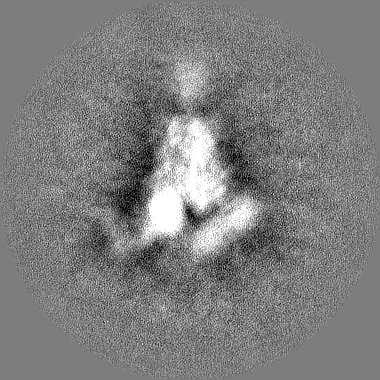
Yorodumi- EMDB-22863: Structure of SARS-CoV spike in complex with CR3022 Fab (Class 3) -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-22863 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Structure of SARS-CoV spike in complex with CR3022 Fab (Class 3) | |||||||||
 Map data Map data | Sharpened map | |||||||||
 Sample Sample |
| |||||||||
| Biological species |   Severe acute respiratory syndrome-related coronavirus Severe acute respiratory syndrome-related coronavirus | |||||||||
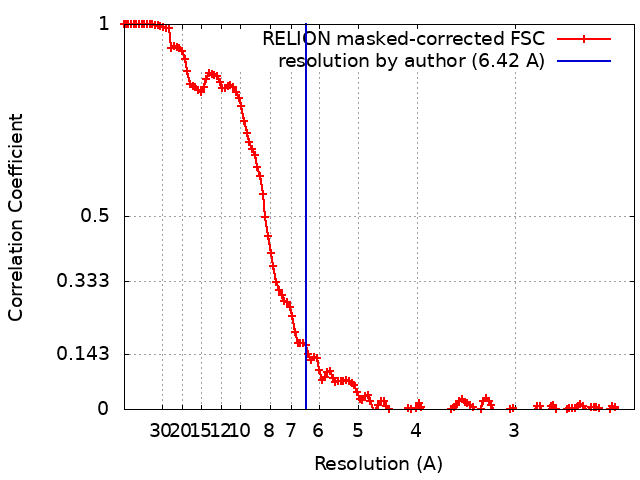
| Method | single particle reconstruction / cryo EM / Resolution: 6.42 Å | |||||||||
 Authors Authors | Ward AB / Bangaru S / Turner HL | |||||||||
| Funding support |  United States, 2 items United States, 2 items
| |||||||||
 Citation Citation | Journal: bioRxiv / Year: 2020 Title: A natural mutation between SARS-CoV-2 and SARS-CoV determines neutralization by a cross-reactive antibody. Authors: Nicholas C Wu / Meng Yuan / Sandhya Bangaru / Deli Huang / Xueyong Zhu / Chang-Chun D Lee / Hannah L Turner / Linghang Peng / Linlin Yang / David Nemazee / Andrew B Ward / Ian A Wilson / Abstract: Epitopes that are conserved among SARS-like coronaviruses are attractive targets for design of cross-reactive vaccines and therapeutics. CR3022 is a SARS-CoV neutralizing antibody to a highly ...Epitopes that are conserved among SARS-like coronaviruses are attractive targets for design of cross-reactive vaccines and therapeutics. CR3022 is a SARS-CoV neutralizing antibody to a highly conserved epitope on the receptor binding domain (RBD) on the spike protein that can cross-react with SARS-CoV-2, but with lower affinity. Using x-ray crystallography, mutagenesis, and binding experiments, we illustrate that of four amino acid differences in the CR3022 epitope between SARS-CoV-2 and SARS-CoV, a single mutation P384A fully determines the affinity difference. CR3022 does not neutralize SARS-CoV-2, but the increased affinity to SARS-CoV-2 P384A mutant now enables neutralization with a similar potency to SARS-CoV. We further investigated CR3022 interaction with the SARS-CoV spike protein by negative-stain EM and cryo-EM. Three CR3022 Fabs bind per trimer with the RBD observed in different up-conformations due to considerable flexibility of the RBD. In one of these conformations, quaternary interactions are made by CR3022 to the N-terminal domain (NTD) of an adjacent subunit. Overall, this study provides insights into antigenic variation and potential for cross-neutralizing epitopes on SARS-like viruses. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
 Movie viewer Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |

- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_22863.map.gz | 195.9 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-22863-v30.xmlemd-22863.xml | 17.6 KB 17.6 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_22863_fsc.xml | 13.6 KB | Display | FSC data file |
| Images |  emd_22863.png emd_22863.png | 42.3 KB | ||
| Masks | emd_22863_msk_1.map | 209.3 MB | Mask map | |
| Others | emd_22863_half_map_1.map.gzemd_22863_half_map_2.map.gz | 166.1 MB 165.9 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-22863ftp://ftp.pdbj.org/pub/emdb/structures/EMD-22863 http://ftp.pdbj.org/pub/emdb/structures/EMD-22863ftp://ftp.pdbj.org/pub/emdb/structures/EMD-22863 | HTTPS FTP |
-Validation report
| Summary document | emd_22863_validation.pdf.gz | 78 KB | Display | EMDB validaton report |
|---|---|---|---|---|
| Full document | emd_22863_full_validation.pdf.gz | 77.1 KB | Display | |
| Data in XML | emd_22863_validation.xml.gz | 494 B | Display | |
| Arichive directory | https://ftp.pdbj.org/pub/emdb/validation_reports/EMD-22863ftp://ftp.pdbj.org/pub/emdb/validation_reports/EMD-22863 | HTTPS FTP |
-Related structure data
| Related structure data | C: citing same article ( |
|---|---|
| Similar structure data |

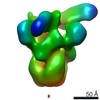
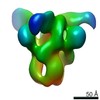








-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_22863.map.gz / Format: CCP4 / Size: 209.3 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Sharpened map | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.15 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















-Supplemental data
-Mask #1
| File | emd_22863_msk_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
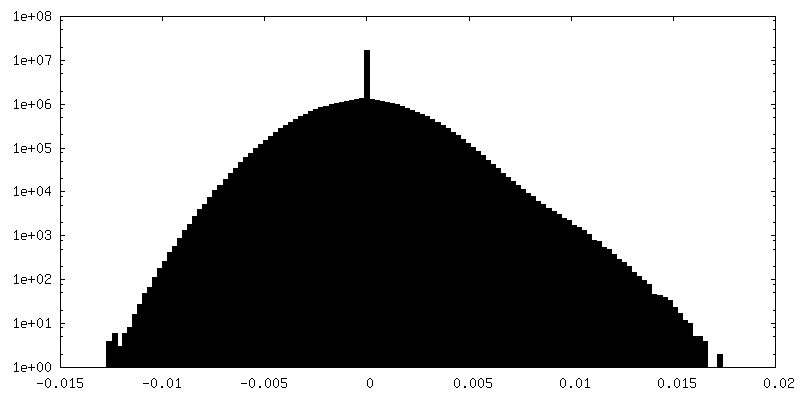
| Density Histograms |






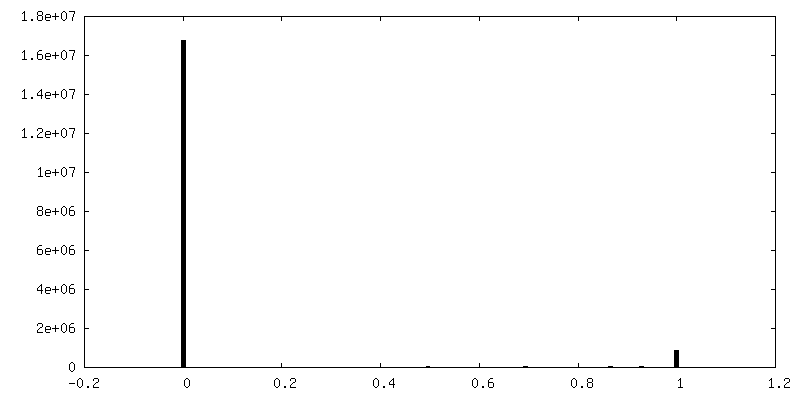
-Half map: Half map 1
| File | emd_22863_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Half map 1 | ||||||||||||
| Projections & Slices |
| ||||||||||||
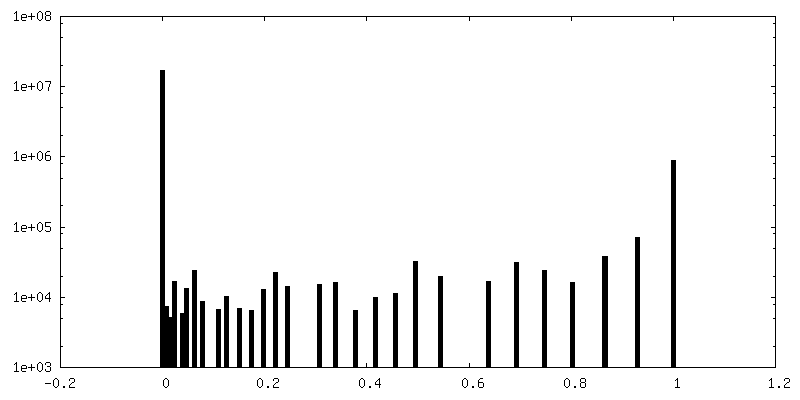
| Density Histograms |
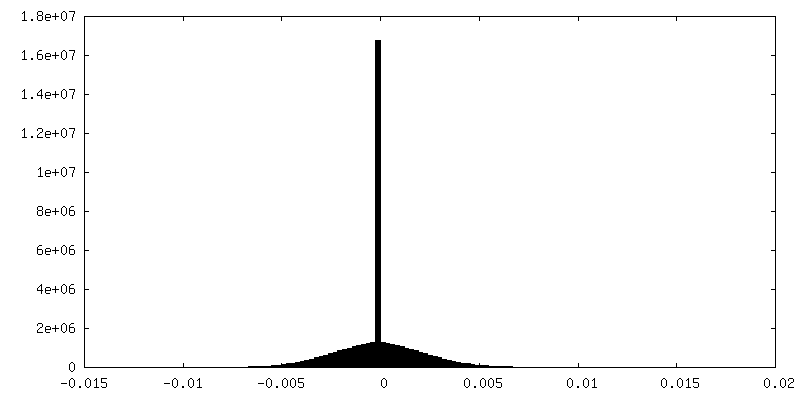

-Half map: Half map 2
| File | emd_22863_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Half map 2 | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |

- Sample components
Sample components
-Entire : Structure of SARS-CoV spike in complex with CR3022 Fab (Class 3)
| Entire | Name: Structure of SARS-CoV spike in complex with CR3022 Fab (Class 3) |
|---|---|
| Components |
|
-Supramolecule #1: Structure of SARS-CoV spike in complex with CR3022 Fab (Class 3)
| Supramolecule | Name: Structure of SARS-CoV spike in complex with CR3022 Fab (Class 3) type: complex / ID: 1 / Parent: 0 / Macromolecule list: all |
|---|---|
| Source (natural) | Organism: Severe acute respiratory syndrome-related coronavirus |
| Recombinant expression | Organism:  Homo sapiens (human) Homo sapiens (human) |
-Macromolecule #1: SARS-CoV spike ectodomain
| Macromolecule | Name: SARS-CoV spike ectodomain / type: protein_or_peptide / ID: 1 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: Severe acute respiratory syndrome-related coronavirus |
| Recombinant expression | Organism: Homo sapiens (human) |
| Sequence | String: MFIFLLFLTL TSGSDLDRCT TFDDVQAPNY TQHTSSMRGV YYPDEIFRSD TLYLTQDLFL PFYSNVTGFH TINHTFGNPV IPFKDGIYFA ATEKSNVVRG WVFGSTMNNK SQSVIIINNS TNVVIRACNF ELCDNPFFAV SKPMGTQTHT MIFDNAFNCT FEYISDAFSL ...String: MFIFLLFLTL TSGSDLDRCT TFDDVQAPNY TQHTSSMRGV YYPDEIFRSD TLYLTQDLFL PFYSNVTGFH TINHTFGNPV IPFKDGIYFA ATEKSNVVRG WVFGSTMNNK SQSVIIINNS TNVVIRACNF ELCDNPFFAV SKPMGTQTHT MIFDNAFNCT FEYISDAFSL DVSEKSGNFK HLREFVFKNK DGFLYVYKGY QPIDVVRDLP SGFNTLKPIF KLPLGINITN FRAILTAFSP AQDIWGTSAA AYFVGYLKPT TFMLKYDENG TITDAVDCSQ NPLAELKCSV KSFEIDKGIY QTSNFRVVPS GDVVRFPNIT NLCPFGEVFN ATKFPSVYAW ERKKISNCVA DYSVLYNSTF FSTFKCYGVS ATKLNDLCFS NVYADSFVVK GDDVRQIAPG QTGVIADYNY KLPDDFMGCV LAWNTRNIDA TSTGNYNYKY RYLRHGKLRP FERDISNVPF SPDGKPCTPP ALNCYWPLND YGFYTTTGIG YQPYRVVVLS FELLNAPATV CGPKLSTDLI KNQCVNFNFN GLTGTGVLTP SSKRFQPFQQ FGRDVSDFTD SVRDPKTSEI LDISPCAFGG VSVITPGTNA SSEVAVLYQD VNCTDVSTAI HADQLTPAWR IYSTGNNVFQ TQAGCLIGAE HVDTSYECDI PIGAGICASY HTVSLLRSTS QKSIVAYTMS LGADSSIAYS NNTIAIPTNF SISITTEVMP VSMAKTSVDC NMYICGDSTE CANLLLQYGS FCTQLNRALS GIAAEQDRNT REVFAQVKQM YKTPTLKYFG GFNFSQILPD PLKPTKRSFI EDLLFNKVTL ADAGFMKQYG ECLGDINARD LICAQKFNGL TVLPPLLTDD MIAAYTAALV SGTATAGWTF GAGAALQIPF AMQMAYRFNG IGVTQNVLYE NQKQIANQFN KAISQIQESL TTTSTALGKL QDVVNQNAQA LNTLVKQLSS NFGAISSVLN DILSRLDPPE AEVQIDRLIT GRLQSLQTYV TQQLIRAAEI RASANLAATK MSECVLGQSK RVDFCGKGYH LMSFPQAAPH GVVFLHVTYV PSQERNFTTA PAICHEGKAY FPREGVFVFN GTSWFITQRN FFSPQIITTD NTFVSGNCDV VIGIINNTVY DPLQPELDSF KEELDKYFKN HTSPDVDLGD ISGINASVVN IQKEIDRLNE VAKNLNESLI DLQELGKYEQ GSGYIPEAPR DGQAYVRKDG EWVLLSTFLG RSLEVLFQGP GHHHHHHHHS AWSHPQFEK |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 0.9 mg/mL |
|---|---|
| Buffer | pH: 7.2 |
| Grid | Model: UltrAuFoil R1.2/1.3 / Material: GOLD / Mesh: 300 / Support film - Material: GOLD / Support film - topology: HOLEY / Pretreatment - Type: PLASMA CLEANING |
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 100 % / Chamber temperature: 283 K / Instrument: FEI VITROBOT MARK IV |
- Electron microscopy
Electron microscopy
| Microscope | FEI TECNAI ARCTICA |
|---|---|
| Image recording | Film or detector model: GATAN K2 SUMMIT (4k x 4k) / Detector mode: COUNTING / Number real images: 2952 / Average exposure time: 11.7 sec. / Average electron dose: 50.0 e/Å2 |
| Electron beam | Acceleration voltage: 200 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | C2 aperture diameter: 70.0 µm / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2.7 mm / Nominal defocus max: 1.6 µm / Nominal defocus min: 0.4 µm / Nominal magnification: 36000 |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER / Cooling holder cryogen: NITROGEN |
| Experimental equipment |  Model: Talos Arctica / Image courtesy: FEI Company |