 ムービー
ムービー コントローラー
コントローラー


+ データを開く
データを開く

- 基本情報
基本情報
| 登録情報 | データベース: PDB / ID: 1ega | ||||||
|---|---|---|---|---|---|---|---|
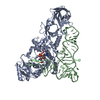
| タイトル | CRYSTAL STRUCTURE OF A WIDELY CONSERVED GTPASE ERA | ||||||
 要素 要素 | PROTEIN (GTP-BINDING PROTEIN ERA) | ||||||
 キーワード キーワード |  HYDROLASE (加水分解酵素) / ERA / GTPASE (GTPアーゼ) / RNA-BINDING / RAS-LIKE HYDROLASE (加水分解酵素) / ERA / GTPASE (GTPアーゼ) / RNA-BINDING / RAS-LIKE | ||||||
| 機能・相同性 |  機能・相同性情報guanosine tetraphosphate binding / ribosomal small subunit binding / ribosomal small subunit biogenesis / small ribosomal subunit rRNA binding / ribosomal small subunit assembly / rRNA binding / protein phosphorylation / GTPase activity / GTP binding / RNA binding ...guanosine tetraphosphate binding / ribosomal small subunit binding / ribosomal small subunit biogenesis / small ribosomal subunit rRNA binding / ribosomal small subunit assembly / rRNA binding / protein phosphorylation / GTPase activity / GTP binding / RNA binding / 細胞膜 / 細胞質基質 / 細胞質 機能・相同性情報guanosine tetraphosphate binding / ribosomal small subunit binding / ribosomal small subunit biogenesis / small ribosomal subunit rRNA binding / ribosomal small subunit assembly / rRNA binding / protein phosphorylation / GTPase activity / GTP binding / RNA binding ...guanosine tetraphosphate binding / ribosomal small subunit binding / ribosomal small subunit biogenesis / small ribosomal subunit rRNA binding / ribosomal small subunit assembly / rRNA binding / protein phosphorylation / GTPase activity / GTP binding / RNA binding / 細胞膜 / 細胞質基質 / 細胞質類似検索 - 分子機能 | ||||||
| 生物種 |  Escherichia coli (大腸菌) Escherichia coli (大腸菌) | ||||||
| 手法 | X線回折 / 多重同系置換 / 解像度: 2.4 Å | ||||||
 データ登録者 データ登録者 | Chen, X. / Ji, X. | ||||||
 引用 引用 | ジャーナル: Proc Natl Acad Sci U S A / 年: 1999 タイトル: Crystal structure of ERA: a GTPase-dependent cell cycle regulator containing an RNA binding motif. 著者: X Chen / D L Court / X Ji /  要旨: ERA forms a unique family of GTPase. It is widely conserved and essential in bacteria. ERA functions in cell cycle control by coupling cell division with growth rate. ERA homologues also are found in ...ERA forms a unique family of GTPase. It is widely conserved and essential in bacteria. ERA functions in cell cycle control by coupling cell division with growth rate. ERA homologues also are found in eukaryotes. Here we report the crystal structure of ERA from Escherichia coli. The structure has been determined at 2.4-A resolution. It reveals a two-domain arrangement of the molecule: an N-terminal domain that resembles p21 Ras and a C-terminal domain that is unique. Structure-based topological search of the C domain fails to reveal any meaningful match, although sequence analysis suggests that it contains a KH domain. KH domains are RNA binding motifs that usually occur in tandem repeats and exhibit low sequence similarity except for the well-conserved segment VIGxxGxxIK. We have identified a betaalphaalphabeta fold that contains the VIGxxGxxIK sequence and is shared by the C domain of ERA and the KH domain. We propose that this betaalphaalphabeta fold is the RNA binding motif, the minimum structural requirement for RNA binding. ERA dimerizes in crystal. The dimer formation involves a significantly distorted switch II region, which may shed light on how ERA protein regulates downstream events. #1: ジャーナル: Mol.Microbiol. / 年: 1998タイトル: Cell Cycle Arrest in Era GTPase Mutants: A Potential Growth Rate-Regulated Checkpoint in E. Coli 著者: Britton, R.A. / Powell, B.S. / Dasgupta, S. / Sun, Q. / Margolin, W. / Lupski, J.R. / Court, D.L. | ||||||
| 履歴 |
|
- 構造の表示
構造の表示
| 構造ビューア | 分子: MolmilJmol/JSmol |
|---|
- ダウンロードとリンク
ダウンロードとリンク
-ダウンロード
| PDBx/mmCIF形式 | 1ega.cif.gz | 128.1 KB | 表示 | PDBx/mmCIF形式 |
|---|---|---|---|---|
| PDB形式 | pdb1ega.ent.gz | 101 KB | 表示 | PDB形式 |
| PDBx/mmJSON形式 | 1ega.json.gz | ツリー表示 | PDBx/mmJSON形式 | |
| その他 |  その他のダウンロード その他のダウンロード |
-検証レポート
| アーカイブディレクトリ | https://data.pdbj.org/pub/pdb/validation_reports/eg/1egaftp://data.pdbj.org/pub/pdb/validation_reports/eg/1ega | HTTPS FTP |
|---|
-関連構造データ







-リンク
 PDBj
PDBj- 集合体
集合体
| 登録構造単位 | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| 単位格子 |
| ||||||||
| 非結晶学的対称性 (NCS) | NCS oper: (Code: given Matrix: (-0.436003, 0.021836, 0.89968), ベクター : |
-要素
| #1: タンパク質 | 分子量: 33858.078 Da / 分子数: 2 / 由来タイプ: 天然 / 由来: (天然) Escherichia coli (大腸菌) / 細胞内の位置: CYTOPLASM/MEMBRANE / 株: TAP106 / 参照: UniProt: P06616#2: 化合物 | ChemComp-SO4 / 硫酸塩  分子量: 96.063 Da / 分子数: 5 / 由来タイプ: 合成 / 式: SO4 分子量: 96.063 Da / 分子数: 5 / 由来タイプ: 合成 / 式: SO4#3: 水 | ChemComp-HOH / | 水 分子量: 18.015 Da / 分子数: 149 / 由来タイプ: 天然 / 式: H2O 分子量: 18.015 Da / 分子数: 149 / 由来タイプ: 天然 / 式: H2O |
|---|
-実験情報
-実験
| 実験 | 手法: X線回折 / 使用した結晶の数: 1 |
|---|
- 試料調製
試料調製
| 結晶 | マシュー密度: 3.5 Å3/Da / 溶媒含有率: 65 % 解説: THE STRUCTURE WAS SOLVED BY A COMBINATION OF MIR AND MAD METHODS. MAD DATA SETS, USING HG ANOMALOUS SCATTERING AT WAVELENGTH 1.00870A, 1.00764A, 0.99184A AND 1.00903A, WERE COLLECTED AT NSLS ...解説: THE STRUCTURE WAS SOLVED BY A COMBINATION OF MIR AND MAD METHODS. MAD DATA SETS, USING HG ANOMALOUS SCATTERING AT WAVELENGTH 1.00870A, 1.00764A, 0.99184A AND 1.00903A, WERE COLLECTED AT NSLS BEAMLINE X9B WITH MAR345 DETECTOR. | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 結晶化 | pH: 8 詳細: PROTEIN 8MG/ML TRIS BUFFER 0.1M, PH 8.0 LITHIUM SULFATE 0.8M SODIUM CHLORIDE 0.8M | ||||||||||||||||||||||||||||||
| 溶液の組成 |
| ||||||||||||||||||||||||||||||
| 結晶化 | *PLUS 手法: 蒸気拡散法, ハンギングドロップ法 | ||||||||||||||||||||||||||||||
| 溶液の組成 | *PLUS
|
-データ収集
| 回折 | 平均測定温度: 100 K |
|---|---|
| 放射光源 | 由来: 回転陽極 / タイプ: RIGAKU / 波長: 1.5418 |
| 検出器 | タイプ: MARRESEARCH / 検出器: IMAGE PLATE / 日付: 1998年6月18日 / 詳細: BENT MIRRORS |
| 放射 | モノクロメーター: GRAPHITE / プロトコル: SINGLE WAVELENGTH / 単色(M)・ラウエ(L): M / 散乱光タイプ: x-ray |
| 放射波長 | 波長: 1.5418 Å / 相対比: 1 |
| 反射 | 解像度: 2.4→20 Å / Num. obs: 35696 / % possible obs: 99.7 % / Observed criterion σ(I): 0 / 冗長度: 6.4 % / Biso Wilson estimate: 53.4 Å2 / Rsym value: 0.056 / Net I/σ(I): 31.8 |
| 反射 シェル | 解像度: 2.4→2.5 Å / 冗長度: 6.1 % / Mean I/σ(I) obs: 3.9 / Rsym value: 0.49 / % possible all: 99.1 |
| 反射 | *PLUS Rmerge(I) obs: 0.056 |
| 反射 シェル | *PLUS % possible obs: 99.1 % / Rmerge(I) obs: 0.49 |
- 解析
解析
| ソフトウェア |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 精密化 | 構造決定の手法: 多重同系置換 / 解像度: 2.4→8 Å / Rfactor Rfree error: 0.0086 / Data cutoff high absF: 100000 / Data cutoff low absF: 0.1 / 交差検証法: THROUGHOUT / σ(F): 4 詳細: N-TERMINAL RESIDUES 1-3 FOR BOTH CHAINS, AND C-TERMINAL RESIDUES 296-301 FOR CHAIN A AND RESIDUES 297-301 FOR CHAIN B WERE NOT OBSERVED IN THE ELECTRON DNESITY AND NOT INCLUDED IN THE MODEL; ...詳細: N-TERMINAL RESIDUES 1-3 FOR BOTH CHAINS, AND C-TERMINAL RESIDUES 296-301 FOR CHAIN A AND RESIDUES 297-301 FOR CHAIN B WERE NOT OBSERVED IN THE ELECTRON DNESITY AND NOT INCLUDED IN THE MODEL; RESIDUES 224 - 228 IN BOTH CHAINS WERE NOT OBSERVED IN THE ELECTRON DENSITY AND NOT INCLUDED IN THE REFINEMENT, BUT BUILT STEREOCHEMICALLY AND INCLUDED IN THE MODEL; RESTRAINTS BETWEEN THE TWO COPIES OF MOLECULES WERE APPLIED DURING THE INITIAL STAGE OF THE REFINEMENT ONLY
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 原子変位パラメータ | Biso mean: 49.5 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 精密化ステップ | サイクル: LAST / 解像度: 2.4→8 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 拘束条件 |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS精密化 シェル | 解像度: 2.4→2.5 Å / Rfactor Rfree error: 0.035 / Total num. of bins used: 8
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ソフトウェア | *PLUS 名称: X-PLOR / バージョン: 3.85 / 分類: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 拘束条件 | *PLUS
|
