 ムービー
ムービー コントローラー
コントローラー


+ データを開く
データを開く

- 基本情報
基本情報
| 登録情報 | 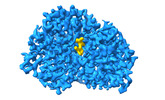 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
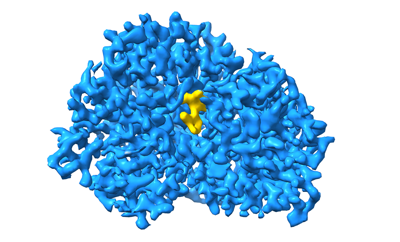
| タイトル | Cryo-EM structure of Maltose Binding Protein | |||||||||
 マップデータ マップデータ | ||||||||||
 試料 試料 |
| |||||||||
 キーワード キーワード |  maltose binding protein / Cryo-EM (低温電子顕微鏡法) / sub-50kDa / atomic resolution. / SUGAR BINDING PROTEIN maltose binding protein / Cryo-EM (低温電子顕微鏡法) / sub-50kDa / atomic resolution. / SUGAR BINDING PROTEIN | |||||||||
| 機能・相同性 |  機能・相同性情報 機能・相同性情報detection of maltose stimulus / maltose binding / maltose transport complex / maltose transport / maltodextrin transmembrane transport / carbohydrate transmembrane transporter activity / ATP-binding cassette (ABC) transporter complex, substrate-binding subunit-containing / carbohydrate transport / ATP-binding cassette (ABC) transporter complex / cell chemotaxis ...detection of maltose stimulus / maltose binding / maltose transport complex / maltose transport / maltodextrin transmembrane transport / carbohydrate transmembrane transporter activity / ATP-binding cassette (ABC) transporter complex, substrate-binding subunit-containing / carbohydrate transport / ATP-binding cassette (ABC) transporter complex / cell chemotaxis / outer membrane-bounded periplasmic space / ペリプラズム / DNA damage response / 生体膜類似検索 - 分子機能 | |||||||||
| 生物種 |  Escherichia coli (大腸菌) Escherichia coli (大腸菌) | |||||||||
| 手法 | 単粒子再構成法 / クライオ電子顕微鏡法 / 解像度: 2.3 Å | |||||||||
 データ登録者 データ登録者 | Yoo Y / Park K / Kim H | |||||||||
| 資金援助 | 1件
| |||||||||
 引用 引用 | ジャーナル: J Mol Biol / 年: 2001 タイトル: Crystal structures of the maltodextrin/maltose-binding protein complexed with reduced oligosaccharides: flexibility of tertiary structure and ligand binding. 著者: X Duan / J A Hall / H Nikaido / F A Quiocho /  要旨: The structure of the maltodextrin or maltose-binding protein, an initial receptor for bacterial ABC-type active transport and chemotaxis, consists of two globular domains that are separated by a ...The structure of the maltodextrin or maltose-binding protein, an initial receptor for bacterial ABC-type active transport and chemotaxis, consists of two globular domains that are separated by a groove wherein the ligand is bound and enclosed by an inter-domain rotation. Here, we report the determination of the crystal structures of the protein complexed with reduced maltooligosaccharides (maltotriitol and maltotetraitol) in both the "closed" and "open" forms. Although these modified sugars bind to the receptor, they are not transported by the wild-type transporter. In the closed structures, the reduced sugars are buried in the groove and bound by both domains, one domain mainly by hydrogen-bonding interactions and the other domain primarily by non-polar interactions with aromatic side-chains. In the open structures, which abrogate both cellular activities of active transport and chemotaxis because of the large separation between the two domains, the sugars are bound almost exclusively to the domain rich in aromatic residues. The binding site for the open chain glucitol residue extends to a subsite that is distinct from those for the glucose residues that were uncovered in prior structural studies of the binding of active linear maltooligosaccharides. Occupation of this subsite may also account for the inability of the reduced oligosaccharides to be transported. The structures reported here, combined with those previously determined for several other complexes with active oligosaccharides in the closed form and with cyclodextrin in the open form, revealed at least four distinct modes of ligand binding but with only one being functionally active. This versatility reflects the flexibility of the protein, from very large motions of interdomain rotation to more localized side-chain conformational changes, and adaptation by the oligosaccharides as well. | |||||||||
| 履歴 |
|
- 構造の表示
構造の表示
| 添付画像 |
|---|
- ダウンロードとリンク
ダウンロードとリンク
-EMDBアーカイブ
| マップデータ | emd_39117.map.gz | 97 MB | EMDBマップデータ形式 | |
|---|---|---|---|---|
| ヘッダ (付随情報) | emd-39117-v30.xmlemd-39117.xml | 15.7 KB 15.7 KB | 表示 表示 | EMDBヘッダ |
| FSC (解像度算出) | emd_39117_fsc.xml | 11.2 KB | 表示 | FSCデータファイル |
| 画像 |  emd_39117.png emd_39117.png | 92.8 KB | ||
| Filedesc metadata | emd-39117.cif.gz | 5.8 KB | ||
| その他 | emd_39117_half_map_1.map.gzemd_39117_half_map_2.map.gz | 95.5 MB 95.5 MB | ||
| アーカイブディレクトリ |  http://ftp.pdbj.org/pub/emdb/structures/EMD-39117ftp://ftp.pdbj.org/pub/emdb/structures/EMD-39117 http://ftp.pdbj.org/pub/emdb/structures/EMD-39117ftp://ftp.pdbj.org/pub/emdb/structures/EMD-39117 | HTTPS FTP |
-関連構造データ

-リンク
| EMDBのページ | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| 「今月の分子」の関連する項目 |



-マップ
| ファイル | ダウンロード / ファイル: emd_39117.map.gz / 形式: CCP4 / 大きさ: 103 MB / タイプ: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ボクセルのサイズ | X=Y=Z: 0.7052 Å | ||||||||||||||||||||
| 密度 |
| ||||||||||||||||||||
| 対称性 | 空間群: 1 | ||||||||||||||||||||
| 詳細 | EMDB XML:
|
-添付データ
-ハーフマップ: #2
| ファイル | emd_39117_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 投影像・断面図 |
| ||||||||||||
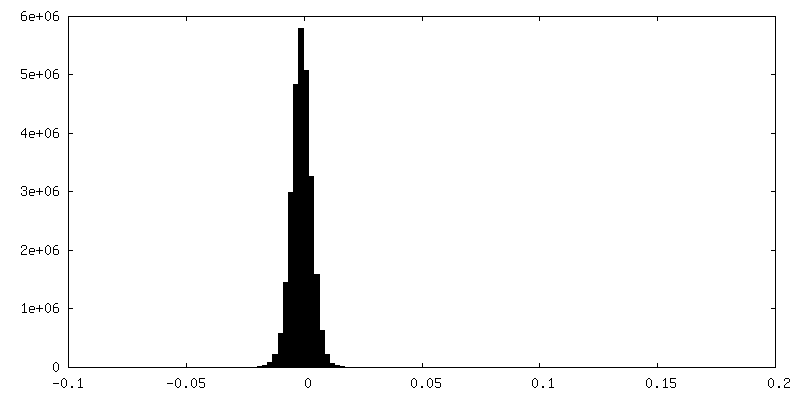
| 密度ヒストグラム |
 Z
Z Y
Y X
X

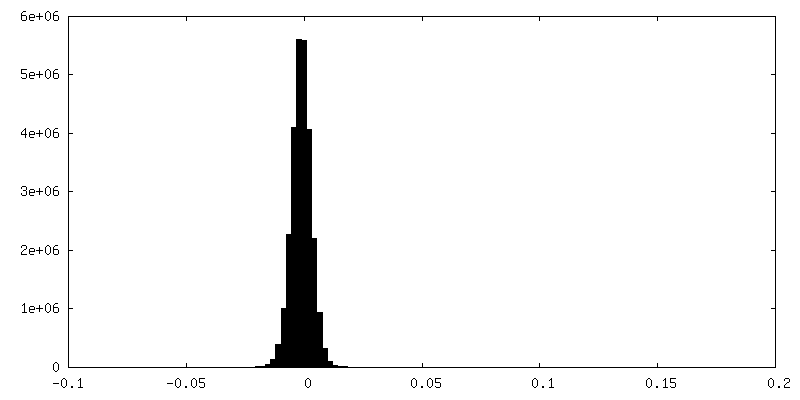
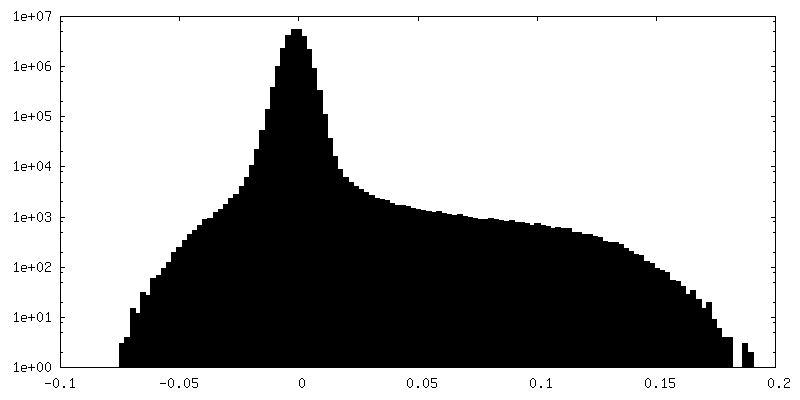
-ハーフマップ: #1
| ファイル | emd_39117_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 投影像・断面図 |
| ||||||||||||
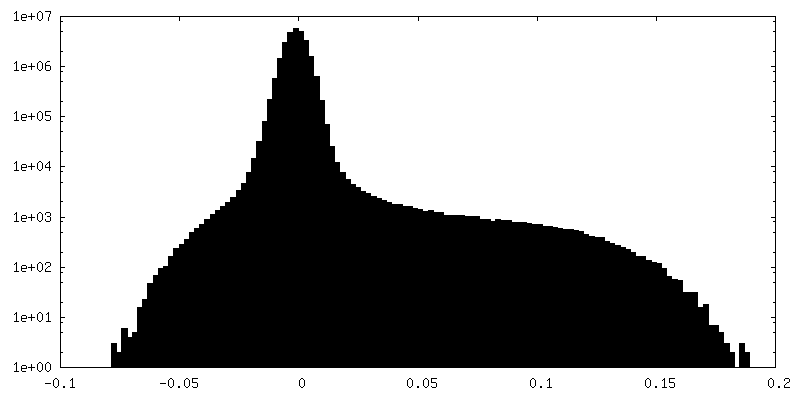
| 密度ヒストグラム |
- 試料の構成要素
試料の構成要素
-全体 : Maltose binding protein monomer
| 全体 | 名称: Maltose binding protein monomer |
|---|---|
| 要素 |
|
-超分子 #1: Maltose binding protein monomer
| 超分子 | 名称: Maltose binding protein monomer / タイプ: complex / ID: 1 / 親要素: 0 / 含まれる分子: #1 |
|---|---|
| 由来(天然) | 生物種: Escherichia coli (大腸菌) |
-分子 #1: Maltose/maltodextrin-binding periplasmic protein
| 分子 | 名称: Maltose/maltodextrin-binding periplasmic protein / タイプ: protein_or_peptide / ID: 1 / コピー数: 1 / 光学異性体: LEVO |
|---|---|
| 由来(天然) | 生物種: Escherichia coli (大腸菌) |
| 分子量 | 理論値: 40.912398 KDa |
| 組換発現 | 生物種: Escherichia coli (大腸菌) |
| 配列 | 文字列: MKIEEGKLVI WINGDKGYNG LAEVGKKFEK DTGIKVTVEH PDKLEEKFPQ VAATGDGPDI IFWAHDRFGG YAQSGLLAEI TPDKAFQDK LYPFTWDAVR YNGKLIAYPI AVEALSLIYN KDLLPNPPKT WEEIPALDKE LKAKGKSALM FNLQEPYFTW P LIAADGGY ...文字列: MKIEEGKLVI WINGDKGYNG LAEVGKKFEK DTGIKVTVEH PDKLEEKFPQ VAATGDGPDI IFWAHDRFGG YAQSGLLAEI TPDKAFQDK LYPFTWDAVR YNGKLIAYPI AVEALSLIYN KDLLPNPPKT WEEIPALDKE LKAKGKSALM FNLQEPYFTW P LIAADGGY AFKYENGKYD IKDVGVDNAG AKAGLTFLVD LIKNKHMNAD TDYSIAEAAF NKGETAMTIN GPWAWSNIDT SK VNYGVTV LPTFKGQPSK PFVGVLSAGI NAASPNKELA KEFLENYLLT DEGLEAVNKD KPLGAVALKS YEEELVKDPR IAA TMENAQ KGEIMPNIPQ MSAFWYAVRT AVINAASGRQ TVDEALKDAQ TRITK UniProtKB: Maltose/maltodextrin-binding periplasmic protein |
-分子 #3: water
| 分子 | 名称: water / タイプ: ligand / ID: 3 / コピー数: 197 / 式: HOH |
|---|---|
| 分子量 | 理論値: 18.015 Da |
| Chemical component information |  ChemComp-HOH: |
-実験情報
-構造解析
| 手法 | クライオ電子顕微鏡法 |
|---|---|
 解析 解析 | 単粒子再構成法 |
| 試料の集合状態 | particle |
-試料調製
| 濃度 | 2 mg/mL |
|---|---|
| 緩衝液 | pH: 7.5 |
| グリッド | モデル: UltrAuFoil R1.2/1.3 / 材質: GOLD / メッシュ: 300 / 前処理 - タイプ: GLOW DISCHARGE / 前処理 - 時間: 60 sec. / 前処理 - 雰囲気: AIR / 前処理 - 気圧: 0.00038 kPa |
| 凍結 | 凍結剤: ETHANE |
- 電子顕微鏡法
電子顕微鏡法
| 顕微鏡 | FEI TITAN KRIOS |
|---|---|
| 電子線 | 加速電圧: 300 kV / 電子線源: FIELD EMISSION GUN |
| 電子光学系 | 照射モード: FLOOD BEAM / 撮影モード: BRIGHT FIELDBright-field microscopy / 最大 デフォーカス(公称値): 1.8 µm / 最小 デフォーカス(公称値): 0.4 µm |
| 撮影 | フィルム・検出器のモデル: FEI FALCON IV (4k x 4k) 平均電子線量: 60.0 e/Å2 |
| 実験機器 |  モデル: Titan Krios / 画像提供: FEI Company |
-画像解析
| 初期モデル | モデルのタイプ: PDB ENTRY PDBモデル - PDB ID: |
|---|---|
| 初期 角度割当 | タイプ: MAXIMUM LIKELIHOOD / ソフトウェア - 名称: cryoSPARC |
| 最終 3次元分類 | クラス数: 3 / ソフトウェア - 名称: cryoSPARC |
| 最終 角度割当 | タイプ: MAXIMUM LIKELIHOOD / ソフトウェア - 名称: cryoSPARC |
| 最終 再構成 | 想定した対称性 - 点群: C1 (非対称) / 解像度のタイプ: BY AUTHOR / 解像度: 2.3 Å / 解像度の算出法: FSC 0.143 CUT-OFF / ソフトウェア - 名称: cryoSPARC / 使用した粒子像数: 517853 |
| FSC曲線 (解像度の算出) | 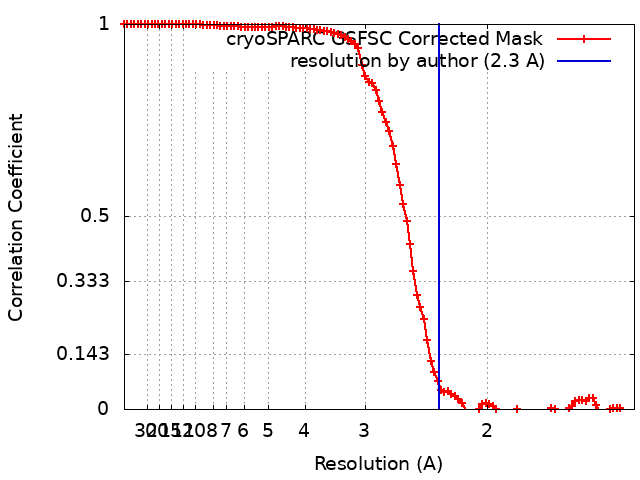 |

