 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 8ybe | ||||||
|---|---|---|---|---|---|---|---|
| Title | Cryo-EM structure of Maltose Binding Protein | ||||||
 Components Components | Maltose/maltodextrin-binding periplasmic protein | ||||||
 Keywords Keywords | SUGAR BINDING PROTEIN /  maltose binding protein / Cryo-EM / sub-50kDa / atomic resolution. maltose binding protein / Cryo-EM / sub-50kDa / atomic resolution. | ||||||
| Function / homology |  Function and homology information Function and homology informationdetection of maltose stimulus / maltose binding / maltose transport complex / maltose transport / maltodextrin transmembrane transport / carbohydrate transmembrane transporter activity / ATP-binding cassette (ABC) transporter complex, substrate-binding subunit-containing / carbohydrate transport / ATP-binding cassette (ABC) transporter complex / cell chemotaxis ...detection of maltose stimulus / maltose binding / maltose transport complex / maltose transport / maltodextrin transmembrane transport / carbohydrate transmembrane transporter activity / ATP-binding cassette (ABC) transporter complex, substrate-binding subunit-containing / carbohydrate transport / ATP-binding cassette (ABC) transporter complex / cell chemotaxis / outer membrane-bounded periplasmic space / periplasmic space / DNA damage response / membraneSimilarity search - Function | ||||||
| Biological species |  Escherichia coli (E. coli) Escherichia coli (E. coli) | ||||||
| Method | ELECTRON MICROSCOPY / single particle reconstruction / cryo EM / Resolution: 2.3 Å | ||||||
 Authors Authors | Yoo, Y. / Park, K. / Kim, H. | ||||||
| Funding support | 1items
| ||||||
 Citation Citation | Journal: To Be Published Title: Atomic resolution structure of MBP using Cryo-EM Authors: Yoo, Y. / Park, K. / Kim, H. #1: Journal: J Mol Biol / Year: 2001Title: Crystal structures of the maltodextrin/maltose-binding protein complexed with reduced oligosaccharides: flexibility of tertiary structure and ligand binding. Authors: X Duan / J A Hall / H Nikaido / F A Quiocho /  Abstract: The structure of the maltodextrin or maltose-binding protein, an initial receptor for bacterial ABC-type active transport and chemotaxis, consists of two globular domains that are separated by a ...The structure of the maltodextrin or maltose-binding protein, an initial receptor for bacterial ABC-type active transport and chemotaxis, consists of two globular domains that are separated by a groove wherein the ligand is bound and enclosed by an inter-domain rotation. Here, we report the determination of the crystal structures of the protein complexed with reduced maltooligosaccharides (maltotriitol and maltotetraitol) in both the "closed" and "open" forms. Although these modified sugars bind to the receptor, they are not transported by the wild-type transporter. In the closed structures, the reduced sugars are buried in the groove and bound by both domains, one domain mainly by hydrogen-bonding interactions and the other domain primarily by non-polar interactions with aromatic side-chains. In the open structures, which abrogate both cellular activities of active transport and chemotaxis because of the large separation between the two domains, the sugars are bound almost exclusively to the domain rich in aromatic residues. The binding site for the open chain glucitol residue extends to a subsite that is distinct from those for the glucose residues that were uncovered in prior structural studies of the binding of active linear maltooligosaccharides. Occupation of this subsite may also account for the inability of the reduced oligosaccharides to be transported. The structures reported here, combined with those previously determined for several other complexes with active oligosaccharides in the closed form and with cyclodextrin in the open form, revealed at least four distinct modes of ligand binding but with only one being functionally active. This versatility reflects the flexibility of the protein, from very large motions of interdomain rotation to more localized side-chain conformational changes, and adaptation by the oligosaccharides as well. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 8ybe.cif.gz | 89.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb8ybe.ent.gz | 63.7 KB | Display | PDB format |
| PDBx/mmJSON format | 8ybe.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/yb/8ybeftp://data.pdbj.org/pub/pdb/validation_reports/yb/8ybe | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  39117MC M: map data used to model this data C: citing same article ( |
|---|---|
| Similar structure data |
-Links
 PDBj
PDBj






- Assembly
Assembly
| Deposited unit | 
|
|---|---|
| 1 |
|
-Components
| #1: Protein | Mass: 40912.398 Da / Num. of mol.: 1 / Mutation: A338V Source method: isolated from a genetically manipulated source Source: (gene. exp.) Escherichia coli (E. coli) / Gene: malE, b4034, JW3994 / Production host: Escherichia coli (E. coli) / References: UniProt: P0AEX9 |
|---|---|
| #2: Polysaccharide | alpha-D-glucopyranose-(1-4)-alpha-D-glucopyranose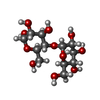  , Oligosaccharide / Class: Nutrient / Mass: 342.297 Da / Num. of mol.: 1 , Oligosaccharide / Class: Nutrient / Mass: 342.297 Da / Num. of mol.: 1Source method: isolated from a genetically manipulated source Details: oligosaccharide / References: alpha-maltose |
| #3: Water | ChemComp-HOH / Water Mass: 18.015 Da / Num. of mol.: 197 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 197 / Source method: isolated from a natural source / Formula: H2O |
| Has ligand of interest | N |
-Experimental details
-Experiment
| Experiment | Method: ELECTRON MICROSCOPY |
|---|---|
| EM experiment | Aggregation state: PARTICLE / 3D reconstruction method: single particle reconstruction |
- Sample preparation
Sample preparation
| Component | Name: Maltose binding protein monomer / Type: COMPLEX / Entity ID: #1 / Source: RECOMBINANT |
|---|---|
| Molecular weight | Experimental value: NO |
| Source (natural) | Organism: Escherichia coli (E. coli) |
| Source (recombinant) | Organism: Escherichia coli (E. coli) |
| Buffer solution | pH: 7.5 |
| Specimen | Conc.: 2 mg/ml / Embedding applied: NO / Shadowing applied: NO / Staining applied: NO / Vitrification applied: YES |
| Specimen support | Grid material: GOLD / Grid mesh size: 300 divisions/in. / Grid type: UltrAuFoil R1.2/1.3 |
| Vitrification | Cryogen name: ETHANE |
- Electron microscopy imaging
Electron microscopy imaging
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
|---|---|
| Microscopy | Model: FEI TITAN KRIOS |
| Electron gun | Electron source: FIELD EMISSION GUN / Accelerating voltage: 300 kV / Illumination mode: FLOOD BEAM |
| Electron lens | Mode: BRIGHT FIELDBright-field microscopy / Nominal defocus max: 1800 nm / Nominal defocus min: 400 nm |
| Image recording | Electron dose: 60 e/Å2 / Film or detector model: FEI FALCON IV (4k x 4k) |
- Processing
Processing
| EM software |
| ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CTF correction | Type: PHASE FLIPPING AND AMPLITUDE CORRECTION | ||||||||||||||||||
| Symmetry | Point symmetry: C1 (asymmetric) | ||||||||||||||||||
| 3D reconstruction | Resolution: 2.3 Å / Resolution method: FSC 0.143 CUT-OFF / Num. of particles: 517853 / Symmetry type: POINT |
