 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-8uja: T33-fn10 - Designed Tetrahedral Protein Cage Using Fragment-based... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 8uja | ||||||
|---|---|---|---|---|---|---|---|
| Title | T33-fn10 - Designed Tetrahedral Protein Cage Using Fragment-based Hydrogen Bond Networks | ||||||
 Components Components |
| ||||||
 Keywords Keywords |  DE NOVO PROTEIN / tetrahedral nanoparticle / designed protein / de novo interface / two-component complex / Rosetta DE NOVO PROTEIN / tetrahedral nanoparticle / designed protein / de novo interface / two-component complex / Rosetta | ||||||
| Function / homology | Enoyl-CoA hydratase, C-terminal / Enoyl-CoA hydratase/isomerase, conserved site / Enoyl-CoA hydratase/isomerase signature. / Enoyl-CoA hydratase/isomerase / Enoyl-CoA hydratase/isomerase / ClpP/crotonase-like domain superfamily / isomerase activity / Enoyl-CoA hydratase/isomerase Function and homology information Function and homology information | ||||||
| Biological species |   Sulfurisphaera tokodaii str. 7 (archaea) Sulfurisphaera tokodaii str. 7 (archaea) Novosphingobium aromaticivorans DSM 12444 (bacteria) Novosphingobium aromaticivorans DSM 12444 (bacteria) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 6 Å | ||||||
 Authors Authors | Meador, K. / Sawaya, M.R. / Yeates, T.O. | ||||||
| Funding support |  United States, 1items United States, 1items
| ||||||
 Citation Citation | Journal: Structure / Year: 2024 Title: A suite of designed protein cages using machine learning and protein fragment-based protocols. Authors: Kyle Meador / Roger Castells-Graells / Roman Aguirre / Michael R Sawaya / Mark A Arbing / Trent Sherman / Chethaka Senarathne / Todd O Yeates / Abstract: Designed protein cages and related materials provide unique opportunities for applications in biotechnology and medicine, but their creation remains challenging. Here, we apply computational ...Designed protein cages and related materials provide unique opportunities for applications in biotechnology and medicine, but their creation remains challenging. Here, we apply computational approaches to design a suite of tetrahedrally symmetric, self-assembling protein cages. For the generation of docked conformations, we emphasize a protein fragment-based approach, while for sequence design of the de novo interface, a comparison of knowledge-based and machine learning protocols highlights the power and increased experimental success achieved using ProteinMPNN. An analysis of design outcomes provides insights for improving interface design protocols, including prioritizing fragment-based motifs, balancing interface hydrophobicity and polarity, and identifying preferred polar contact patterns. In all, we report five structures for seven protein cages, along with two structures of intermediate assemblies, with the highest resolution reaching 2.0 Å using cryo-EM. This set of designed cages adds substantially to the body of available protein nanoparticles, and to methodologies for their creation. #1: Journal: bioRxiv / Year: 2023Title: A Suite of Designed Protein Cages Using Machine Learning Algorithms and Protein Fragment-Based Protocols Authors: Meador, K. / Castells-Graells, R. / Aguirre, R. / Sawaya, M.R. / Arbing, M.A. / Sherman, T. / Senarathne, C. / Yeates, T.O. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 8uja.cif.gz | 1.1 MB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb8uja.ent.gz | 931.4 KB | Display | PDB format |
| PDBx/mmJSON format | 8uja.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/uj/8ujaftp://data.pdbj.org/pub/pdb/validation_reports/uj/8uja | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  8uf0C  8ui2C  8ukmC  8umpC  8umrC  8un1C C: citing same article ( |
|---|---|
| Similar structure data |

-Links
 PDBj
PDBj

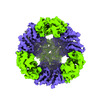
- Assembly
Assembly
| Deposited unit | 
| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||||||
| 2 | 
| ||||||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 12122.230 Da / Num. of mol.: 8 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Sulfurisphaera tokodaii str. 7 (archaea)Production host: Escherichia coli BL21(DE3) (bacteria) / Variant (production host): LOBSTR#2: Protein | Mass: 27671.732 Da / Num. of mol.: 8 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Novosphingobium aromaticivorans DSM 12444 (bacteria)Gene: Saro_3457 / Production host: Escherichia coli BL21(DE3) (bacteria) / Variant (production host): LOBSTR / References: UniProt: A4XEF6 |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.01 Å3/Da / Density % sol: 59.09 % |
|---|---|
| Crystal grow | Temperature: 294 K / Method: vapor diffusion, hanging drop / pH: 7.5 Details: 100 mM HEPES pH 7.5, 10% PEG 8000, 10% Ethylene glycol |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS / Beamline: 24-ID-C / Wavelength: 0.97918 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: DECTRIS EIGER2 X 16M / Detector: PIXEL / Date: Apr 1, 2022 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Monochromator: Si(111) / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 0.97918 Å / Relative weight: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 6→92.11 Å / Num. obs: 18590 / % possible obs: 100 % / Redundancy: 10.6 % / Biso Wilson estimate: 356.46 Å2 / CC1/2: 0.997 / Rmerge(I) obs: 0.174 / Rrim(I) all: 0.182 / Net I/σ(I): 9.17 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell |
|
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 6→92.11 Å / SU ML: 0.8147 / Cross valid method: FREE R-VALUE / σ(F): 1.37 / Phase error: 31.5139 Stereochemistry target values: GeoStd + Monomer Library + CDL v1.2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.1 Å / Solvent model: FLAT BULK SOLVENT MODEL | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 400.92 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 6→92.11 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Origin x: 80.4415769715 Å / Origin y: 45.1882430276 Å / Origin z: 43.114529597 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group | Selection details: all |
