 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-7tdp: Structure of Paenibacillus polymyxa GS bound to Met-Sox-P-ADP (Tr... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 7tdp | ||||||
|---|---|---|---|---|---|---|---|
| Title | Structure of Paenibacillus polymyxa GS bound to Met-Sox-P-ADP (Transition state complex) to 1.98 Angstom | ||||||
 Components Components | Glutamine synthetase | ||||||
 Keywords Keywords | LIGASE / glutamate-ammonium ligase / GlnR / GS / feedback inhibition / transcription coregulator / glnRA | ||||||
| Function / homology |  Function and homology informationglutamine synthetase / glutamine biosynthetic process / glutamine synthetase activity / ATP binding / metal ion binding / cytoplasm Function and homology informationglutamine synthetase / glutamine biosynthetic process / glutamine synthetase activity / ATP binding / metal ion binding / cytoplasmSimilarity search - Function | ||||||
| Biological species |  Paenibacillus polymyxa (bacteria) Paenibacillus polymyxa (bacteria) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 1.98 Å | ||||||
 Authors Authors | Schumacher, M.A. | ||||||
| Funding support |  United States, 1items United States, 1items
| ||||||
 Citation Citation | Journal: Nat Commun / Year: 2022 Title: Molecular dissection of the glutamine synthetase-GlnR nitrogen regulatory circuitry in Gram-positive bacteria. Authors: Brady A Travis / Jared V Peck / Raul Salinas / Brandon Dopkins / Nicholas Lent / Viet D Nguyen / Mario J Borgnia / Richard G Brennan / Maria A Schumacher / Abstract: How bacteria sense and respond to nitrogen levels are central questions in microbial physiology. In Gram-positive bacteria, nitrogen homeostasis is controlled by an operon encoding glutamine ...How bacteria sense and respond to nitrogen levels are central questions in microbial physiology. In Gram-positive bacteria, nitrogen homeostasis is controlled by an operon encoding glutamine synthetase (GS), a dodecameric machine that assimilates ammonium into glutamine, and the GlnR repressor. GlnR detects nitrogen excess indirectly by binding glutamine-feedback-inhibited-GS (FBI-GS), which activates its transcription-repression function. The molecular mechanisms behind this regulatory circuitry, however, are unknown. Here we describe biochemical and structural analyses of GS and FBI-GS-GlnR complexes from pathogenic and non-pathogenic Gram-positive bacteria. The structures show FBI-GS binds the GlnR C-terminal domain within its active-site cavity, juxtaposing two GlnR monomers to form a DNA-binding-competent GlnR dimer. The FBI-GS-GlnR interaction stabilizes the inactive GS conformation. Strikingly, this interaction also favors a remarkable dodecamer to tetradecamer transition in some GS, breaking the paradigm that all bacterial GS are dodecamers. These data thus unveil unique structural mechanisms of transcription and enzymatic regulation. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 7tdp.cif.gz | 556 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb7tdp.ent.gz | 454.6 KB | Display | PDB format |
| PDBx/mmJSON format | 7tdp.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/td/7tdpftp://data.pdbj.org/pub/pdb/validation_reports/td/7tdp | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  7tdvC  7teaC  7tecC  7tenC  7tf6C  7tf7C  7tf9C  7tfaC  7tfbC  7tfcC  7tfdC  7tfeC  4lniS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |








-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 50338.988 Da / Num. of mol.: 3 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Paenibacillus polymyxa (bacteria) / Gene: glnA2, LK13_01575, NCTC10343_02989 / Production host: Escherichia coli (E. coli) / References: UniProt: A0A0F0G8G2, glutamine synthetase#2: Chemical | Adenosine diphosphate  Mass: 427.201 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C10H15N5O10P2 / Comment: ADP, energy-carrying molecule*YM Mass: 427.201 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C10H15N5O10P2 / Comment: ADP, energy-carrying molecule*YM#3: Chemical | 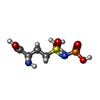  Mass: 260.205 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C5H13N2O6PS / Feature type: SUBJECT OF INVESTIGATION Mass: 260.205 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C5H13N2O6PS / Feature type: SUBJECT OF INVESTIGATION#4: Chemical | ChemComp-MG /   Mass: 24.305 Da / Num. of mol.: 9 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 9 / Source method: obtained synthetically / Formula: Mg#5: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 799 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 799 / Source method: isolated from a natural source / Formula: H2OHas ligand of interest | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.15 Å3/Da / Density % sol: 60.97 % |
|---|---|
| Crystal grow | Temperature: 278 K / Method: vapor diffusion, hanging drop / Details: 30% PEG mono methyl ether 2000, 0.1 M Tris pH 8.0 |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ALS / Beamline: 5.0.2 / Wavelength: 1.08 Å |
| Detector | Type: DECTRIS PILATUS 2M-F / Detector: PIXEL / Date: Sep 12, 2021 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.08 Å / Relative weight: 1 |
| Reflection | Resolution: 1.98→48.7 Å / Num. obs: 132443 / % possible obs: 98.5 % / Redundancy: 13.1 % / CC1/2: 0.999 / Rpim(I) all: 0.03 / Rsym value: 0.108 / Net I/σ(I): 15.4 |
| Reflection shell | Resolution: 1.98→2.05 Å / Redundancy: 4.6 % / Mean I/σ(I) obs: 1.4 / Num. unique obs: 12010 / CC1/2: 0.486 / Rpim(I) all: 0.544 / Rsym value: 1.154 |
-Phasing
| Phasing | Method: molecular replacement |
|---|
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 4LNI Resolution: 1.98→48.7 Å / SU ML: 0.25 / Cross valid method: THROUGHOUT / σ(F): 1.33 / Phase error: 21.31 / Stereochemistry target values: ML
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 111.37 Å2 / Biso mean: 42.1516 Å2 / Biso min: 20.44 Å2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 1.98→48.7 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Rfactor Rfree error: 0 / Total num. of bins used: 14
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Origin x: 17.1311 Å / Origin y: 17.0315 Å / Origin z: 41.6451 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
