 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 7svm | ||||||
|---|---|---|---|---|---|---|---|
| Title | DPP8 IN COMPLEX WITH LIGAND ICeD-2 | ||||||
 Components Components | Dipeptidyl peptidase 8 | ||||||
 Keywords Keywords | HYDROLASE / DIPEPTIDYL PEPTIDASE | ||||||
| Function / homology |  Function and homology informationdipeptidyl-peptidase IV / dipeptidyl-peptidase activity / negative regulation of programmed cell death / aminopeptidase activity / serine-type peptidase activity / immune response / apoptotic process / proteolysis / cytosol / cytoplasm Function and homology informationdipeptidyl-peptidase IV / dipeptidyl-peptidase activity / negative regulation of programmed cell death / aminopeptidase activity / serine-type peptidase activity / immune response / apoptotic process / proteolysis / cytosol / cytoplasmSimilarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.69 Å | ||||||
 Authors Authors | Lammens, A. / Hollenstein, K. / Klein, D.J. | ||||||
| Funding support | 1items
| ||||||
 Citation Citation | Journal: Acs Chem.Biol. / Year: 2022 Title: A Phenotypic Screen Identifies Potent DPP9 Inhibitors Capable of Killing HIV-1 Infected Cells. Authors: Moore, K.P. / Schwaid, A.G. / Tudor, M. / Park, S. / Beshore, D.C. / Converso, A. / Shipe, W.D. / Anand, R. / Lan, P. / Moningka, R. / Rothman, D.M. / Sun, W. / Chi, A. / Cornella-Taracido, ...Authors: Moore, K.P. / Schwaid, A.G. / Tudor, M. / Park, S. / Beshore, D.C. / Converso, A. / Shipe, W.D. / Anand, R. / Lan, P. / Moningka, R. / Rothman, D.M. / Sun, W. / Chi, A. / Cornella-Taracido, I. / Adam, G.C. / Bahnck-Teets, C. / Carroll, S.S. / Fay, J.F. / Goh, S.L. / Lusen, J. / Quan, S. / Rodriguez, S. / Xu, M. / Andrews, C.L. / Song, C. / Filzen, T. / Li, J. / Hollenstein, K. / Klein, D.J. / Lammens, A. / Lim, U.M. / Fang, Z. / McHale, C. / Li, Y. / Lu, M. / Diamond, T.L. / Howell, B.J. / Zuck, P. / Balibar, C.J. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 7svm.cif.gz | 1 MB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb7svm.ent.gz | 879.5 KB | Display | PDB format |
| PDBx/mmJSON format | 7svm.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/sv/7svmftp://data.pdbj.org/pub/pdb/validation_reports/sv/7svm | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  7svlC  7svnC  7svoC  6eooS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |
-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| |||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| |||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | 
| |||||||||||||||||||||||||||||||||||||||||||||||||||||
| Unit cell |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS domain:
NCS domain segments: Component-ID: 0 / Beg auth comp-ID: LYS / Beg label comp-ID: LYS / End auth comp-ID: ILE / End label comp-ID: ILE / Refine code: 0 / Auth seq-ID: 47 - 898 / Label seq-ID: 47 - 898
NCS ensembles :
|
-Components
| #1: Protein | / DP8 / Dipeptidyl peptidase IV-related protein 1 / DPRP-1 / Dipeptidyl peptidase VIII / DPP VIII / ...DP8 / Dipeptidyl peptidase IV-related protein 1 / DPRP-1 / Dipeptidyl peptidase VIII / DPP VIII / Prolyl dipeptidase DPP8 Mass: 103483.352 Da / Num. of mol.: 3 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: DPP8, DPRP1, MSTP097, MSTP135, MSTP141 / Production host:   Spodoptera frugiperda (fall armyworm) / References: UniProt: Q6V1X1, dipeptidyl-peptidase IV Spodoptera frugiperda (fall armyworm) / References: UniProt: Q6V1X1, dipeptidyl-peptidase IV#2: Chemical | ChemComp-D06 / (   Mass: 327.464 Da / Num. of mol.: 13 / Source method: obtained synthetically / Formula: C20H29N3O / Feature type: SUBJECT OF INVESTIGATION Mass: 327.464 Da / Num. of mol.: 13 / Source method: obtained synthetically / Formula: C20H29N3O / Feature type: SUBJECT OF INVESTIGATION#3: Chemical | ChemComp-TMO / Trimethylamine N-oxide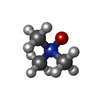  Mass: 75.110 Da / Num. of mol.: 14 / Source method: obtained synthetically / Formula: C3H9NO Mass: 75.110 Da / Num. of mol.: 14 / Source method: obtained synthetically / Formula: C3H9NO#4: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 473 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 473 / Source method: isolated from a natural source / Formula: H2OHas ligand of interest | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 4.52 Å3/Da / Density % sol: 72.8 % |
|---|---|
| Crystal grow | Temperature: 277 K / Method: vapor diffusion / Details: 0.46 M NaCitrate pH 6.75 |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SLS  / Beamline: X10SA / Wavelength: 0.9998 Å / Beamline: X10SA / Wavelength: 0.9998 Å |
| Detector | Type: DECTRIS PILATUS 6M / Detector: PIXEL / Date: Feb 3, 2019 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9998 Å / Relative weight: 1 |
| Reflection | Resolution: 2.69→135.29 Å / Num. obs: 138235 / % possible obs: 96.1 % / Redundancy: 3 % / Rmerge(I) obs: 0.064 / Net I/σ(I): 1.97 |
| Reflection shell | Resolution: 2.69→2.94 Å / Redundancy: 2.9 % / Rmerge(I) obs: 0.58 / Num. unique obs: 32528 / % possible all: 97.3 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 6EOO Resolution: 2.69→135.29 Å / Cor.coef. Fo:Fc: 0.96 / Cor.coef. Fo:Fc free: 0.945 / SU B: 21.15 / SU ML: 0.202 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.308 / ESU R Free: 0.233 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: U VALUES : WITH TLS ADDED HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 202.15 Å2 / Biso mean: 64.883 Å2 / Biso min: 23.58 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 2.69→135.29 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints NCS | Refine-ID: X-RAY DIFFRACTION / Type: interatomic distance / Weight position: 0.05
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.69→2.76 Å / Rfactor Rfree error: 0 / Total num. of bins used: 20
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
