 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-6t4b: CRYSTAL STRUCTURE OF HUMAN TDP-43 N-TERMINAL DOMAIN AT 2.55 A RES... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6t4b | ||||||
|---|---|---|---|---|---|---|---|
| Title | CRYSTAL STRUCTURE OF HUMAN TDP-43 N-TERMINAL DOMAIN AT 2.55 A RESOLUTION | ||||||
 Components Components | TAR DNA-binding protein 43 | ||||||
 Keywords Keywords | DNA BINDING PROTEIN / MND / NTD domain / TDP-43 | ||||||
| Function / homology |  Function and homology information Function and homology informationnuclear inner membrane organization / interchromatin granule / perichromatin fibrils / 3'-UTR-mediated mRNA destabilization / 3'-UTR-mediated mRNA stabilization / intracellular non-membrane-bounded organelle / negative regulation by host of viral transcription / pre-mRNA intronic binding / response to endoplasmic reticulum stress / RNA splicing ...nuclear inner membrane organization / interchromatin granule / perichromatin fibrils / 3'-UTR-mediated mRNA destabilization / 3'-UTR-mediated mRNA stabilization / intracellular non-membrane-bounded organelle / negative regulation by host of viral transcription / pre-mRNA intronic binding / response to endoplasmic reticulum stress / RNA splicing / negative regulation of protein phosphorylation / molecular condensate scaffold activity / mRNA 3'-UTR binding / regulation of protein stability / regulation of circadian rhythm / positive regulation of insulin secretion / mRNA processing / cytoplasmic stress granule / positive regulation of protein import into nucleus / rhythmic process / regulation of gene expression / double-stranded DNA binding / regulation of apoptotic process / amyloid fibril formation / regulation of cell cycle / nuclear speck / RNA polymerase II cis-regulatory region sequence-specific DNA binding / negative regulation of gene expression / lipid binding / mitochondrion / DNA binding / RNA binding / nucleoplasm / identical protein binding / nucleusSimilarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.55 Å | ||||||
 Authors Authors | Watanabe, T.F. / Wright, G.S.A. / Amporndanai, K. / Antonyuk, S.V. / Hasnain, S.S. | ||||||
 Citation Citation | Journal: Iscience / Year: 2020 Title: Purification and Structural Characterization of Aggregation-Prone Human TDP-43 Involved in Neurodegenerative Diseases. Authors: Wright, G.S.A. / Watanabe, T.F. / Amporndanai, K. / Plotkin, S.S. / Cashman, N.R. / Antonyuk, S.V. / Hasnain, S.S. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6t4b.cif.gz | 93.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6t4b.ent.gz | 71.4 KB | Display | PDB format |
| PDBx/mmJSON format | 6t4b.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/t4/6t4bftp://data.pdbj.org/pub/pdb/validation_reports/t4/6t4b | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  5mdiS S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 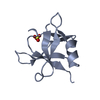
| ||||||||
| 2 | 
| ||||||||
| 3 | 
| ||||||||
| 4 | 
| ||||||||
| 5 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | / TDP-43 Mass: 8875.903 Da / Num. of mol.: 5 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: TARDBP, TDP43 / Production host:  Escherichia coli (E. coli) / Variant (production host): BL52 / References: UniProt: Q13148 Escherichia coli (E. coli) / Variant (production host): BL52 / References: UniProt: Q13148#2: Chemical | ChemComp-SO4 / Sulfate  Mass: 96.063 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: SO4#3: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 233 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 233 / Source method: isolated from a natural source / Formula: H2OHas ligand of interest | N | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3 Å3/Da / Density % sol: 59 % / Description: needle like |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, hanging drop / pH: 6.5 Details: 0.2M Sodium bromide, 0.1M Bis-Tris propane 6.5, 20% PEG 3350 |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: Diamond  / Beamline: I03 / Wavelength: 0.9763 Å / Beamline: I03 / Wavelength: 0.9763 Å |
| Detector | Type: DECTRIS PILATUS 6M / Detector: PIXEL / Date: Jul 16, 2018 |
| Radiation | Monochromator: Si111 / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9763 Å / Relative weight: 1 |
| Reflection | Resolution: 2.55→78.9 Å / Num. obs: 16549 / % possible obs: 98.7 % / Redundancy: 3.9 % / Biso Wilson estimate: 38.3 Å2 / CC1/2: 0.98 / Rmerge(I) obs: 0.156 / Net I/σ(I): 5.7 |
| Reflection shell | Resolution: 2.55→2.62 Å / Redundancy: 4 % / Rmerge(I) obs: 0.986 / Mean I/σ(I) obs: 1.6 / Num. unique obs: 1212 / CC1/2: 0.549 / % possible all: 99.1 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 5mdi Resolution: 2.55→78.9 Å / Cor.coef. Fo:Fc: 0.936 / Cor.coef. Fo:Fc free: 0.913 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.582 / ESU R Free: 0.307 Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS U VALUES : REFINED INDIVIDUALLY
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 140.39 Å2 / Biso mean: 46.148 Å2 / Biso min: 18 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 2.55→78.9 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.55→2.616 Å / Total num. of bins used: 20
|
