Entry Database : PDB / ID : 6t2wTitle Crystal structure of the CSF1R kinase domain with a dihydropurinone inhibitor (compound 4) Macrophage colony-stimulating factor 1 receptor Keywords / / Function / homology Function Domain/homology Component
/ / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / Biological species Homo sapiens (human)Method / / / / Resolution : 1.7 Å Authors Schimpl, M. / Goldberg, F.W. / Finlay, M.R.V. / Ting, A.K.T. / Beattie, D. / Lamont, G.M. / Fallan, C. / Wrigley, G.L. / Howard, M.R. / Williamson, B. ...Schimpl, M. / Goldberg, F.W. / Finlay, M.R.V. / Ting, A.K.T. / Beattie, D. / Lamont, G.M. / Fallan, C. / Wrigley, G.L. / Howard, M.R. / Williamson, B. / Davies, B.R. / Cadogan, E.B. / Ramos-Montoya, A. / Dean, E. Journal : J.Med.Chem. / Year : 2020Title: The Discovery of 7-Methyl-2-[(7-methyl[1,2,4]triazolo[1,5-a]pyridin-6-yl)amino]-9-(tetrahydro-2H-pyran-4-yl)-7,9-dihydro-8H-purin-8-one (AZD7648), a Potent and Selective DNA-Dependent Protein ... Title : The Discovery of 7-Methyl-2-[(7-methyl[1,2,4]triazolo[1,5-a]pyridin-6-yl)amino]-9-(tetrahydro-2H-pyran-4-yl)-7,9-dihydro-8H-purin-8-one (AZD7648), a Potent and Selective DNA-Dependent Protein Kinase (DNA-PK) Inhibitor.Authors: Goldberg, F.W. / Finlay, M.R.V. / Ting, A.K.T. / Beattie, D. / Lamont, G.M. / Fallan, C. / Wrigley, G.L. / Schimpl, M. / Howard, M.R. / Williamson, B. / Vazquez-Chantada, M. / Barratt, D.G. ... Authors : Goldberg, F.W. / Finlay, M.R.V. / Ting, A.K.T. / Beattie, D. / Lamont, G.M. / Fallan, C. / Wrigley, G.L. / Schimpl, M. / Howard, M.R. / Williamson, B. / Vazquez-Chantada, M. / Barratt, D.G. / Davies, B.R. / Cadogan, E.B. / Ramos-Montoya, A. / Dean, E. History Deposition Oct 9, 2019 Deposition site / Processing site Revision 1.0 Jan 1, 2020 Provider / Type Revision 1.1 Jan 22, 2020 Group / Category / citation_authorItem / _citation.year / _citation_author.nameRevision 1.2 Apr 22, 2020 Group / Category / citation_authorItem _citation.journal_volume / _citation.page_first ... _citation.journal_volume / _citation.page_first / _citation.page_last / _citation_author.identifier_ORCID Revision 1.3 May 1, 2024 Group / Database references / Refinement descriptionCategory chem_comp_atom / chem_comp_bond ... chem_comp_atom / chem_comp_bond / database_2 / pdbx_initial_refinement_model Item / _database_2.pdbx_database_accession
Show all Show less
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords TRANSFERASE / kinase fold / type I kinase inhibitor
TRANSFERASE / kinase fold / type I kinase inhibitor Function and homology information
Function and homology information
 Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads











 PDBj
PDBj














 Assembly
Assembly


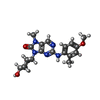
 Mass: 383.444 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C20H25N5O3 / Feature type: SUBJECT OF INVESTIGATION
Mass: 383.444 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C20H25N5O3 / Feature type: SUBJECT OF INVESTIGATION
 Mass: 96.063 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: SO4
Mass: 96.063 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: SO4 Mass: 18.015 Da / Num. of mol.: 234 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 234 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: I03 / Wavelength: 0.97626 Å
/ Beamline: I03 / Wavelength: 0.97626 Å Processing
Processing