| 登録情報 | データベース: PDB / ID: 6o1x
|
|---|
| タイトル | Structure of pCW3 conjugation coupling protein TcpA monomer form with ATPgS |
|---|
 要素 要素 | DNA translocase coupling protein |
|---|
 キーワード キーワード |  TRANSLOCASE (輸送酵素) / ATPase (ATPアーゼ) / Conjugation TRANSLOCASE (輸送酵素) / ATPase (ATPアーゼ) / Conjugation |
|---|
| 機能・相同性 |  機能・相同性情報 機能・相同性情報 |
|---|
| 生物種 |   Clostridium perfringens (ウェルシュ菌) Clostridium perfringens (ウェルシュ菌) |
|---|
| 手法 | X線回折 / シンクロトロン / 分子置換 / 解像度: 2.46 Å |
|---|
 データ登録者 データ登録者 | Traore, D.A.K. / Ahktar, N. / Torres, V.T. / Adams, V. / Coulibaly, F. / Panjikar, S. / Caradoc-Davies, T.T. / Rood, J.I. / Whisstock, J.C. |
|---|
| 資金援助 |  オーストラリア, 1件 オーストラリア, 1件 | 組織 | 認可番号 | 国 |
|---|
| National Health and Medical Research Council (NHMRC, Australia) | GNT1104502 | オーストラリア |
|
|---|
 引用 引用 | ジャーナル: To be published
タイトル: Structure of pCW3 conjugation coupling protein TcpA
著者: Traore, D.A.K. / Whisstock, J.C. |
|---|
| 履歴 | | 登録 | 2019年2月22日 | 登録サイト: RCSB / 処理サイト: RCSB |
|---|
| 改定 1.0 | 2020年2月26日 | Provider: repository / タイプ: Initial release |
|---|
| 改定 2.0 | 2020年7月29日 | Group: Atomic model / Data collection ...Atomic model / Data collection / Derived calculations / Structure summary
カテゴリ: atom_site / atom_site_anisotrop ...atom_site / atom_site_anisotrop / chem_comp / entity / pdbx_chem_comp_identifier / pdbx_entity_nonpoly / struct_site / struct_site_gen
Item: _atom_site.auth_atom_id / _atom_site.label_atom_id ..._atom_site.auth_atom_id / _atom_site.label_atom_id / _atom_site_anisotrop.pdbx_auth_atom_id / _atom_site_anisotrop.pdbx_label_atom_id / _chem_comp.name / _entity.pdbx_description / _pdbx_entity_nonpoly.name
解説: Carbohydrate remediation / Provider: repository / タイプ: Remediation |
|---|
| 改定 2.1 | 2023年10月11日 | Group: Data collection / Database references ...Data collection / Database references / Refinement description / Structure summary
カテゴリ: chem_comp / chem_comp_atom ...chem_comp / chem_comp_atom / chem_comp_bond / database_2 / pdbx_initial_refinement_model
Item: _chem_comp.pdbx_synonyms / _database_2.pdbx_DOI / _database_2.pdbx_database_accession |
|---|
|
|---|
 ムービー
ムービー コントローラー
コントローラー


 データを開く
データを開く
 基本情報
基本情報 構造の表示
構造の表示 ダウンロードとリンク
ダウンロードとリンク その他のダウンロード
その他のダウンロード












 PDBj
PDBj



 集合体
集合体


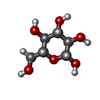
 タイプ: D-saccharide, beta linking / 分子量: 180.156 Da / 分子数: 2 / 由来タイプ: 組換発現 / 式: C6H12O6
タイプ: D-saccharide, beta linking / 分子量: 180.156 Da / 分子数: 2 / 由来タイプ: 組換発現 / 式: C6H12O6
 分子量: 523.247 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C10H16N5O12P3S
分子量: 523.247 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C10H16N5O12P3S 分子量: 18.015 Da / 分子数: 184 / 由来タイプ: 天然 / 式: H2O
分子量: 18.015 Da / 分子数: 184 / 由来タイプ: 天然 / 式: H2O 試料調製
試料調製 解析
解析