 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6mlt | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of the V. cholerae biofilm matrix protein Bap1 | ||||||
 Components Components | Hemolysin-related protein | ||||||
 Keywords Keywords | SUGAR BINDING PROTEIN / biofilm matrix protein / carbohydrate-binding protein /  metal-binding protein / secreted protein metal-binding protein / secreted protein | ||||||
| Function / homology | Hemolysin, beta-prism lectin / Beta-prism lectin / Jacalin-like lectin domain superfamily / EF-Hand 1, calcium-binding site / EF-hand calcium-binding domain. / CITRATE ANION / Hemolysin-related protein Function and homology information Function and homology information | ||||||
| Biological species |  Vibrio cholerae serotype O1 (bacteria) Vibrio cholerae serotype O1 (bacteria) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / SIRAS / Resolution: 1.9 Å | ||||||
 Authors Authors | Kaus, K. / Biester, A. / Chupp, E. / Lu, K. / Vidsudharomn, C. / Olson, R. | ||||||
 Citation Citation | Journal: J.Biol.Chem. / Year: 2019 Title: The 1.9 angstrom crystal structure of the extracellular matrix protein Bap1 fromVibrio choleraeprovides insights into bacterial biofilm adhesion. Authors: Kaus, K. / Biester, A. / Chupp, E. / Lu, J. / Visudharomn, C. / Olson, R. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6mlt.cif.gz | 343.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6mlt.ent.gz | 280.7 KB | Display | PDB format |
| PDBx/mmJSON format | 6mlt.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ml/6mltftp://data.pdbj.org/pub/pdb/validation_reports/ml/6mlt | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|










-Links
 PDBj
PDBj





- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
-Protein , 1 types, 1 molecules A
| #1: Protein | Mass: 66237.812 Da / Num. of mol.: 1 / Mutation: Y415-K471 Deletion Source method: isolated from a genetically manipulated source Details: Loop including residues Y415-K471 deleted genetically Source: (gene. exp.) Vibrio cholerae serotype O1 (bacteria) / Strain: ATCC 39315 / El Tor Inaba N16961 / Gene: VC_1888 / Plasmid: pNGFP-BC / Details (production host): N-terminal GFP fusion / Production host: Escherichia coli (E. coli) / Strain (production host): BL21 / Variant (production host): NEB T7 Express / References: UniProt: Q9KQW0 |
|---|
-Non-polymers , 5 types, 481 molecules 

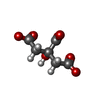


| #2: Chemical |  Mass: 40.078 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Ca Mass: 40.078 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Ca#3: Chemical | ChemComp-NA /  Mass: 22.990 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: Na Mass: 22.990 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: Na#4: Chemical | ChemComp-FLC / | Citric acid Mass: 189.100 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H5O7 Mass: 189.100 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H5O7#5: Chemical | Glycerol Mass: 92.094 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C3H8O3#6: Water | ChemComp-HOH / | WaterMass: 18.015 Da / Num. of mol.: 471 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.93 Å3/Da / Density % sol: 57.97 % |
|---|---|
| Crystal grow | Temperature: 295 K / Method: vapor diffusion, hanging drop / pH: 7.5 / Details: 0.1M sodium citrate, 10% PEG 3350 |
-Data collection
| Diffraction | Mean temperature: 100 K | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: NSLS-II  / Beamline: 17-ID-1 / Wavelength: 1.25414 Å / Beamline: 17-ID-1 / Wavelength: 1.25414 Å | ||||||||||||||||||||||||
| Detector | Type: DECTRIS EIGER X 9M / Detector: PIXEL / Date: Jul 21, 2017 | ||||||||||||||||||||||||
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 1.25414 Å / Relative weight: 1 | ||||||||||||||||||||||||
| Reflection | Resolution: 1.9→47.64 Å / Num. obs: 62568 / % possible obs: 99.7 % / Redundancy: 11.1 % / CC1/2: 0.998 / Rmerge(I) obs: 0.114 / Rpim(I) all: 0.034 / Rrim(I) all: 0.12 / Net I/σ(I): 13.8 | ||||||||||||||||||||||||
| Reflection shell | Diffraction-ID: 1
|
-Phasing
| Phasing | Method: SIRAS |
|---|
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: SIRAS / Resolution: 1.9→47.64 Å / SU ML: 0.22 / Cross valid method: THROUGHOUT / σ(F): 1.02 / Phase error: 15.76
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 132.95 Å2 / Biso mean: 30.2647 Å2 / Biso min: 13.76 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 1.9→47.64 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Rfactor Rfree error: 0 / % reflection Rfree: 5 %
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
