 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-6fm9: Crystal structure of human UDP-N-acetylglucosamine-dolichyl-phosp... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6fm9 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of human UDP-N-acetylglucosamine-dolichyl-phosphate N-acetylglucosaminephosphotransferase (DPAGT1) | ||||||
 Components Components | UDP-N-acetylglucosamine--dolichyl-phosphate N-acetylglucosaminephosphotransferase | ||||||
 Keywords Keywords |  TRANSFERASE / Protein glycosylation / integral membrane protein / congenital myasthenic syndrome 13 / Structural Genomics / Structural Genomics Consortium / SGC TRANSFERASE / Protein glycosylation / integral membrane protein / congenital myasthenic syndrome 13 / Structural Genomics / Structural Genomics Consortium / SGC | ||||||
| Function / homology |  Function and homology information Function and homology informationBiosynthesis of the N-glycan precursor (dolichol lipid-linked oligosaccharide, LLO) and transfer to a nascent protein / Defective DPAGT1 causes CDG-1j, CMSTA2 / UDP-N-acetylglucosamine-dolichyl-phosphate N-acetylglucosaminephosphotransferase / UDP-N-acetylglucosamine-dolichyl-phosphate N-acetylglucosaminephosphotransferase activity / UDP-N-acetylglucosamine-lysosomal-enzyme N-acetylglucosaminephosphotransferase activity / dolichol-linked oligosaccharide biosynthetic process / UDP-N-acetylglucosamine metabolic process / protein N-linked glycosylation / glycosyltransferase activity / intracellular membrane-bounded organelle ...Biosynthesis of the N-glycan precursor (dolichol lipid-linked oligosaccharide, LLO) and transfer to a nascent protein / Defective DPAGT1 causes CDG-1j, CMSTA2 / UDP-N-acetylglucosamine-dolichyl-phosphate N-acetylglucosaminephosphotransferase / UDP-N-acetylglucosamine-dolichyl-phosphate N-acetylglucosaminephosphotransferase activity / UDP-N-acetylglucosamine-lysosomal-enzyme N-acetylglucosaminephosphotransferase activity / dolichol-linked oligosaccharide biosynthetic process / UDP-N-acetylglucosamine metabolic process / protein N-linked glycosylation / glycosyltransferase activity / intracellular membrane-bounded organelle / endoplasmic reticulum membrane / membrane / identical protein binding / metal ion bindingSimilarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 3.6 Å | ||||||
 Authors Authors | Pike, A.C.W. / Dong, Y.Y. / Chu, A. / Tessitore, A. / Goubin, S. / Dong, L. / Mukhopadhyay, S. / Mahajan, P. / Chalk, R. / Berridge, G. ...Pike, A.C.W. / Dong, Y.Y. / Chu, A. / Tessitore, A. / Goubin, S. / Dong, L. / Mukhopadhyay, S. / Mahajan, P. / Chalk, R. / Berridge, G. / Wang, D. / Kupinska, K. / Belaya, K. / Beeson, D. / Burgess-Brown, N. / Edwards, A.M. / Arrowsmith, C.H. / Bountra, C. / Carpenter, E.P. / Structural Genomics Consortium (SGC) | ||||||
| Funding support |  United Kingdom, 1items United Kingdom, 1items
| ||||||
 Citation Citation | Journal: Cell / Year: 2018 Title: Structures of DPAGT1 Explain Glycosylation Disease Mechanisms and Advance TB Antibiotic Design. Authors: Dong, Y.Y. / Wang, H. / Pike, A.C.W. / Cochrane, S.A. / Hamedzadeh, S. / Wyszynski, F.J. / Bushell, S.R. / Royer, S.F. / Widdick, D.A. / Sajid, A. / Boshoff, H.I. / Park, Y. / Lucas, R. / ...Authors: Dong, Y.Y. / Wang, H. / Pike, A.C.W. / Cochrane, S.A. / Hamedzadeh, S. / Wyszynski, F.J. / Bushell, S.R. / Royer, S.F. / Widdick, D.A. / Sajid, A. / Boshoff, H.I. / Park, Y. / Lucas, R. / Liu, W.M. / Lee, S.S. / Machida, T. / Minall, L. / Mehmood, S. / Belaya, K. / Liu, W.W. / Chu, A. / Shrestha, L. / Mukhopadhyay, S.M.M. / Strain-Damerell, C. / Chalk, R. / Burgess-Brown, N.A. / Bibb, M.J. / Barry Iii, C.E. / Robinson, C.V. / Beeson, D. / Davis, B.G. / Carpenter, E.P. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6fm9.cif.gz | 163.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6fm9.ent.gz | 130.1 KB | Display | PDB format |
| PDBx/mmJSON format | 6fm9.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/fm/6fm9ftp://data.pdbj.org/pub/pdb/validation_reports/fm/6fm9 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  5levC  5o5eC  6fwzC  4j72S S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 46215.797 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: DPAGT1, DPAGT2 / Plasmid: PFB-LIC-BSE / Production host:   Spodoptera frugiperda (fall armyworm) Spodoptera frugiperda (fall armyworm)References: UniProt: Q9H3H5, UDP-N-acetylglucosamine-dolichyl-phosphate N-acetylglucosaminephosphotransferase |
|---|---|
| #2: Chemical | ChemComp-P6L / (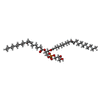  Mass: 746.991 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C40H75O10P / Comment: phospholipid*YM Mass: 746.991 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C40H75O10P / Comment: phospholipid*YM |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 4.06 Å3/Da / Density % sol: 69.72 % |
|---|---|
| Crystal grow | Temperature: 277 K / Method: vapor diffusion, sitting drop / pH: 6.5 / Details: 0.05M ADA pH 6.5 -- 24% PEG400 |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: Diamond / Beamline: I04-1 / Wavelength: 0.91741 Å |
| Detector | Type: DECTRIS PILATUS3 6M / Detector: PIXEL / Date: Jan 26, 2015 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.91741 Å / Relative weight: 1 |
| Reflection | Resolution: 3.6→72.06 Å / Num. obs: 9509 / % possible obs: 99.6 % / Redundancy: 6.2 % / Biso Wilson estimate: 99.7 Å2 / CC1/2: 0.999 / Rmerge(I) obs: 0.055 / Rpim(I) all: 0.024 / Rrim(I) all: 0.061 / Net I/σ(I): 17.8 |
| Reflection shell | Resolution: 3.6→3.69 Å / Redundancy: 6.6 % / Rmerge(I) obs: 1.132 / Mean I/σ(I) obs: 1.8 / Num. unique obs: 681 / CC1/2: 0.666 / Rpim(I) all: 0.475 / Rrim(I) all: 1.23 / % possible all: 100 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 4J72 Resolution: 3.6→30 Å / Cor.coef. Fo:Fc: 0.874 / Cor.coef. Fo:Fc free: 0.912 / Cross valid method: THROUGHOUT / σ(F): 0 / SU Rfree Blow DPI: 0.538 Details: Refined using 5LEV as target reference restraints. Single TLS group for entire protein chain.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 171.41 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.51 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: 1 / Resolution: 3.6→30 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 3.6→4.03 Å / Total num. of bins used: 5
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Origin x: 47.2354 Å / Origin y: 50.461 Å / Origin z: 129.432 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group | Selection details: { A|7 - A|405 } |
