National Institutes of Health/National Institute of Diabetes and Digestive and Kidney Disease (NIH/NIDDK)
DK042303
United States
National Institutes of Health/National Cancer Institute (NIH/NCI)
CA108874
United States
National Institutes of Health/National Institute of General Medical Sciences (NIH/NIGMS)
GM008270
United States
Citation
Journal: ACS Chem Biol / Year: 2018 Title: Biosynthesis of t-Butyl in Apratoxin A: Functional Analysis and Architecture of a PKS Loading Module. Authors: Meredith A Skiba / Andrew P Sikkema / Nathan A Moss / Andrew N Lowell / Min Su / Rebecca M Sturgis / Lena Gerwick / William H Gerwick / David H Sherman / Janet L Smith / Abstract: The unusual feature of a t-butyl group is found in several marine-derived natural products including apratoxin A, a Sec61 inhibitor produced by the cyanobacterium Moorea bouillonii PNG 5-198. Here, ...The unusual feature of a t-butyl group is found in several marine-derived natural products including apratoxin A, a Sec61 inhibitor produced by the cyanobacterium Moorea bouillonii PNG 5-198. Here, we determine that the apratoxin A t-butyl group is formed as a pivaloyl acyl carrier protein (ACP) by AprA, the polyketide synthase (PKS) loading module of the apratoxin A biosynthetic pathway. AprA contains an inactive "pseudo" GCN5-related N-acetyltransferase domain (ΨGNAT) flanked by two methyltransferase domains (MT1 and MT2) that differ distinctly in sequence. Structural, biochemical, and precursor incorporation studies reveal that MT2 catalyzes unusually coupled decarboxylation and methylation reactions to transform dimethylmalonyl-ACP, the product of MT1, to pivaloyl-ACP. Further, pivaloyl-ACP synthesis is primed by the fatty acid synthase malonyl acyltransferase (FabD), which compensates for the ΨGNAT and provides the initial acyl-transfer step to form AprA malonyl-ACP. Additionally, images of AprA from negative stain electron microscopy reveal multiple conformations that may facilitate the individual catalytic steps of the multienzyme module.
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords TRANSFERASE /
TRANSFERASE /  Function and homology information
Function and homology information Moorea bouillonii (bacteria)
Moorea bouillonii (bacteria) Authors
Authors United States, 3items
United States, 3items  Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads












 PDBj
PDBj





 Assembly
Assembly

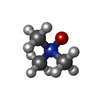
 Mass: 75.110 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H9NO
Mass: 75.110 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H9NO
 Type: L-peptide linking / Mass: 384.411 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C14H20N6O5S / Feature type: SUBJECT OF INVESTIGATION
Type: L-peptide linking / Mass: 384.411 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C14H20N6O5S / Feature type: SUBJECT OF INVESTIGATION Mass: 18.015 Da / Num. of mol.: 125 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 125 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation Processing
Processing