- PDB-6a71: Crystal Structure of Human ATP7B and TM Complex -
+
Open data
ID or keywords:
Loading...
-
Basic information
Entry
Database: PDB / ID: 6a71
Title
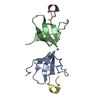
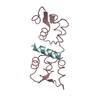
Crystal Structure of Human ATP7B and TM Complex
Components
ATP7B proteinWilson disease protein
Keywords
METAL TRANSPORT / Copper transporter protein
Function / homology
Function and homology information
regulation of biological quality / protein maturation by copper ion transfer / P-type divalent copper transporter activity / P-type monovalent copper transporter activity / P-type Cu+ transporter / copper ion export / copper ion import / copper ion transmembrane transporter activity / sequestering of calcium ion / copper ion transport ...regulation of biological quality / protein maturation by copper ion transfer / P-type divalent copper transporter activity / P-type monovalent copper transporter activity / P-type Cu+ transporter / copper ion export / copper ion import / copper ion transmembrane transporter activity / sequestering of calcium ion / copper ion transport / xenobiotic detoxification by transmembrane export across the plasma membrane / intracellular zinc ion homeostasis / response to copper ion / Ion transport by P-type ATPases / intracellular copper ion homeostasis / lactation / monoatomic ion transmembrane transport / trans-Golgi network membrane / establishment of localization in cell / trans-Golgi network / late endosome / copper ion binding / Golgi membrane / Golgi apparatus / ATP hydrolysis activity / mitochondrion / ATP binding / membrane / plasma membrane Similarity search - Function
Resolution: 1.6→43.87 Å / Cor.coef. Fo:Fc: 0.968 / Cor.coef. Fo:Fc free: 0.956 / SU B: 1.505 / SU ML: 0.053 / Cross valid method: THROUGHOUT / ESU R: 0.082 / ESU R Free: 0.083 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
Rfactor
Num. reflection
% reflection
Selection details
Rfree
0.19537
1029
4.9 %
RANDOM
Rwork
0.16521
-
-
-
obs
0.16676
19928
97.76 %
-
Solvent computation
Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Wilson disease protein
Wilson disease protein  Keywords
Keywords Function and homology information
Function and homology information
 Authors
Authors China, 1items
China, 1items  Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads









 PDBj
PDBj
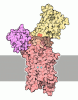




 Assembly
Assembly





 Mass: 106.120 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H10O3
Mass: 106.120 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H10O3 Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3
Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca
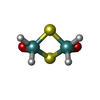
Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca Mass: 292.041 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: H4Mo2O2S2
Mass: 292.041 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: H4Mo2O2S2 Sample preparation
Sample preparation Processing
Processing