 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
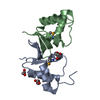
| Entry | Database: PDB / ID: 5zo6 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of C166, a backbone circularized G-CSF | ||||||
 Components Components | Granulocyte colony-stimulating factor | ||||||
 Keywords Keywords | CYTOKINE / four-helix bundle / back-bone circularization | ||||||
| Function / homology |  Function and homology informationgranulocyte colony-stimulating factor receptor binding / granulocyte colony-stimulating factor signaling pathway / positive regulation of myeloid cell differentiation / granulocyte differentiation / regulation of actin filament organization / Other interleukin signaling / positive regulation of actin filament polymerization / cellular response to cytokine stimulus / Interleukin-10 signaling / Signaling by CSF3 (G-CSF) ...granulocyte colony-stimulating factor receptor binding / granulocyte colony-stimulating factor signaling pathway / positive regulation of myeloid cell differentiation / granulocyte differentiation / regulation of actin filament organization / Other interleukin signaling / positive regulation of actin filament polymerization / cellular response to cytokine stimulus / Interleukin-10 signaling / Signaling by CSF3 (G-CSF) / endocytic vesicle lumen / lysosomal lumen / cytokine activity / growth factor activity / Inactivation of CSF3 (G-CSF) signaling / cytokine-mediated signaling pathway / positive regulation of peptidyl-tyrosine phosphorylation / response to ethanol / cellular response to lipopolysaccharide / positive regulation of phosphatidylinositol 3-kinase/protein kinase B signal transduction / immune response / positive regulation of cell population proliferation / enzyme binding / positive regulation of transcription by RNA polymerase II / extracellular space / extracellular region Function and homology informationgranulocyte colony-stimulating factor receptor binding / granulocyte colony-stimulating factor signaling pathway / positive regulation of myeloid cell differentiation / granulocyte differentiation / regulation of actin filament organization / Other interleukin signaling / positive regulation of actin filament polymerization / cellular response to cytokine stimulus / Interleukin-10 signaling / Signaling by CSF3 (G-CSF) ...granulocyte colony-stimulating factor receptor binding / granulocyte colony-stimulating factor signaling pathway / positive regulation of myeloid cell differentiation / granulocyte differentiation / regulation of actin filament organization / Other interleukin signaling / positive regulation of actin filament polymerization / cellular response to cytokine stimulus / Interleukin-10 signaling / Signaling by CSF3 (G-CSF) / endocytic vesicle lumen / lysosomal lumen / cytokine activity / growth factor activity / Inactivation of CSF3 (G-CSF) signaling / cytokine-mediated signaling pathway / positive regulation of peptidyl-tyrosine phosphorylation / response to ethanol / cellular response to lipopolysaccharide / positive regulation of phosphatidylinositol 3-kinase/protein kinase B signal transduction / immune response / positive regulation of cell population proliferation / enzyme binding / positive regulation of transcription by RNA polymerase II / extracellular space / extracellular regionSimilarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.7 Å | ||||||
 Authors Authors | Shibuya, R. / Miyafusa, T. / Honda, S. | ||||||
| Funding support |  Japan, 1items Japan, 1items
| ||||||
 Citation Citation | Journal: Febs J. / Year: 2019 Title: Stabilization of backbone-circularized protein is attained by synergistic gains in enthalpy of folded structure and entropy of unfolded structure. Authors: Shibuya, R. / Miyafusa, T. / Honda, S. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 5zo6.cif.gz | 48.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb5zo6.ent.gz | 32.3 KB | Display | PDB format |
| PDBx/mmJSON format | 5zo6.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/zo/5zo6ftp://data.pdbj.org/pub/pdb/validation_reports/zo/5zo6 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1bgcS S: Starting model for refinement |
|---|---|
| Similar structure data |









-Links
 PDBj
PDBj










- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | / G-CSF / Pluripoietin Mass: 17893.605 Da / Num. of mol.: 1 / Fragment: UNP residues 37-205 / Mutation: C11S, A166G Source method: isolated from a genetically manipulated source Details: 166th Gly and 1st Ser are connected with a peptide bond Source: (gene. exp.) Homo sapiens (human) / Gene: CSF3, C17orf33, GCSF / Production host:  Escherichia coli (E. coli) / References: UniProt: P09919 Escherichia coli (E. coli) / References: UniProt: P09919 |
|---|---|
| #2: Water | ChemComp-HOH / Water Mass: 18.015 Da / Num. of mol.: 105 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 105 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 1.74 Å3/Da / Density % sol: 29.34 % |
|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, hanging drop / pH: 5.2 Details: sodium acetate trihydrate, sodium chloride dehydrate, PEG 1500, MPD |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: Photon Factory / Beamline: AR-NW12A / Wavelength: 1 Å |
| Detector | Type: ADSC QUANTUM 270 / Detector: CCD / Date: Mar 11, 2016 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 1.7→47.15 Å / Num. obs: 14235 / % possible obs: 99.7 % / Redundancy: 6.9 % / Rmerge(I) obs: 0.06 / Net I/σ(I): 21.8 |
| Reflection shell | Resolution: 1.7→1.73 Å / Redundancy: 7.1 % / Rmerge(I) obs: 0.271 / Mean I/σ(I) obs: 7.3 / Num. unique obs: 747 / % possible all: 99.5 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 1BGC Resolution: 1.7→36.17 Å / Cor.coef. Fo:Fc: 0.958 / Cor.coef. Fo:Fc free: 0.932 / SU B: 2.18 / SU ML: 0.074 / Cross valid method: THROUGHOUT / ESU R: 0.12 / ESU R Free: 0.12 / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 20.73 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: 1 / Resolution: 1.7→36.17 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|