Mass: 18.015 Da / Num. of mol.: 69 / Source method: isolated from a natural source / Formula: H2O
Sequence details
COMPLETE SEQUENCE OF UPAIN-2-3W3A PEPTIDE IS CSA(7XC)GLENHAAC. THIS PEPTIDE WAS HYDROLYZED BY ...COMPLETE SEQUENCE OF UPAIN-2-3W3A PEPTIDE IS CSA(7XC)GLENHAAC. THIS PEPTIDE WAS HYDROLYZED BY SERINE PROTEASE (UPA), ONLY N-TERMINAL RESIDUES CSA(7XC) AND C-TERMINAL RESIDUES AC WERE OBSERVED. MEANWHILE, N-TERMINAL CYS AND C-TERMINAL CYS LINKED TOGETHER THROUGH SSBOND.
-
Experimental details
-
Experiment
Experiment
Method: X-RAY DIFFRACTION / Number of used crystals: 1
-
Sample preparation
Crystal
Density Matthews: 2.04 Å3/Da / Density % sol: 39.67 %
Crystal grow
Temperature: 298 K / Method: vapor diffusion, sitting drop Details: 50mM sodium citrate, pH 4.6, 2.0M ammonium sulfate supplemented with 5% polyethylene glycol 400
Resolution: 1.75→39.23 Å / Cor.coef. Fo:Fc: 0.956 / Cor.coef. Fo:Fc free: 0.942 / SU B: 3.44 / SU ML: 0.109 / Cross valid method: THROUGHOUT / ESU R: 0.154 / ESU R Free: 0.14 / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
Rfactor
Num. reflection
% reflection
Selection details
Rfree
0.249
1146
5.1 %
RANDOM
Rwork
0.213
-
-
-
obs
0.215
21226
96.8 %
-
Solvent computation
Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Serine proteases / uPA / pepetide inhibitors / HYDROLASE-HYDROLASE INHIBITOR complex
Serine proteases / uPA / pepetide inhibitors / HYDROLASE-HYDROLASE INHIBITOR complex Function and homology information
Function and homology information
 Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads
















 PDBj
PDBj



 Assembly
Assembly

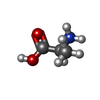
 Type: L-peptide linking / Mass: 89.093 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H7NO2
Type: L-peptide linking / Mass: 89.093 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H7NO2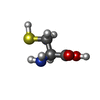
 Type: L-peptide linking / Mass: 121.158 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H7NO2S
Type: L-peptide linking / Mass: 121.158 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H7NO2S Mass: 18.015 Da / Num. of mol.: 69 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 69 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: BL17U / Wavelength: 0.979 Å
/ Beamline: BL17U / Wavelength: 0.979 Å Processing
Processing