 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-5k1z: Joint X-ray/neutron structure of MTAN complex with p-ClPh-Thio-DA... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 5k1z | ||||||
|---|---|---|---|---|---|---|---|
| Title | Joint X-ray/neutron structure of MTAN complex with p-ClPh-Thio-DADMe-ImmA | ||||||
 Components Components | Aminodeoxyfutalosine nucleosidase | ||||||
 Keywords Keywords |  HYDROLASE / Neutron / Nucleosidase / Joint Neutron and X-ray / Helicobacter pylori HYDROLASE / Neutron / Nucleosidase / Joint Neutron and X-ray / Helicobacter pylori | ||||||
| Function / homology |  Function and homology information Function and homology informationaminodeoxyfutalosine nucleosidase / 6-amino-6-deoxyfutalosine hydrolase activity / adenosylhomocysteine nucleosidase / adenosylhomocysteine nucleosidase activity / methylthioadenosine nucleosidase activity / L-methionine salvage from S-adenosylmethionine / nucleoside catabolic process / L-methionine salvage from methylthioadenosine / menaquinone biosynthetic process / cytosolSimilarity search - Function | ||||||
| Biological species |   Helicobacter pylori (bacteria) Helicobacter pylori (bacteria) | ||||||
| Method | NEUTRON DIFFRACTION / X-RAY DIFFRACTION / NUCLEAR REACTOR / Resolution: 2.6 Å | ||||||
 Authors Authors | Banco, M.T. / Kovalevsky, A.Y. / Ronning, D.R. | ||||||
 Citation Citation | Journal: Proc. Natl. Acad. Sci. U.S.A. / Year: 2016 Title: Neutron structures of the Helicobacter pylori 5'-methylthioadenosine nucleosidase highlight proton sharing and protonation states. Authors: Banco, M.T. / Mishra, V. / Ostermann, A. / Schrader, T.E. / Evans, G.B. / Kovalevsky, A. / Ronning, D.R. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 5k1z.cif.gz | 106.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb5k1z.ent.gz | 83.5 KB | Display | PDB format |
| PDBx/mmJSON format | 5k1z.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/k1/5k1zftp://data.pdbj.org/pub/pdb/validation_reports/k1/5k1z | HTTPS FTP |
|---|
-Related structure data


















-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
| ||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | Mass: 24918.703 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Helicobacter pylori (bacteria) / Gene: mtnN, mtn, jhp_0082 / Production host: Escherichia coli (E. coli)References: UniProt: Q9ZMY2, aminodeoxyfutalosine nucleosidase, adenosylhomocysteine nucleosidase |
|---|---|
| #2: Chemical | ChemComp-4CT / (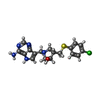  Mass: 389.902 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C18H20ClN5OS Mass: 389.902 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C18H20ClN5OS |
| #3: Chemical | ChemComp-DOD / Heavy water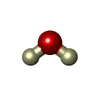  Mass: 18.015 Da / Num. of mol.: 58 / Source method: isolated from a natural source / Formula: D2O Mass: 18.015 Da / Num. of mol.: 58 / Source method: isolated from a natural source / Formula: D2O |
-Experimental details
-Experiment
| Experiment |
|
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.71 Å3/Da / Density % sol: 54.63 % |
|---|---|
| Crystal grow | Temperature: 277.15 K / Method: evaporation / pH: 7 Details: 100 mM HEPES, pH 7.0, 28-30% w/v PEG550 MME, 50 mM magnesium chloride hexahydrate |
-Data collection
| Diffraction |
| ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source |
| ||||||||||||||||||
| Detector |
| ||||||||||||||||||
| Radiation |
| ||||||||||||||||||
| Radiation wavelength |
| ||||||||||||||||||
| Reflection | Resolution: 2.6→31.88 Å / Num. obs: 6426 / % possible obs: 75.1 % / Redundancy: 3.1 % / Net I/σ(I): 3.3 | ||||||||||||||||||
| Reflection shell | Resolution: 2.25→2.35 Å / Redundancy: 4.1 % / Rmerge(I) obs: 0.532 / Mean I/σ(I) obs: 2.5 / % possible all: 100 |

- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Biso max: 94.37 Å2 / Biso mean: 41.83 Å2 / Biso min: 9.71 Å2 / Cross valid method: FREE R-VALUE / Bsol: 31.7033 Å2 / ksol: 0.43874 e/Å3
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine funct minimized |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.6→31.79 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Total num. of bins used: 8
|
