 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-5jlt: The crystal structure of the bacteriophage T4 MotA C-terminal dom... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 5jlt | ||||||
|---|---|---|---|---|---|---|---|
| Title | The crystal structure of the bacteriophage T4 MotA C-terminal domain in complex with dsDNA reveals a novel protein-DNA recognition motif | ||||||
 Components Components |
| ||||||
 Keywords Keywords |  VIRAL PROTEIN/DNA / MotA / dsDNA / "Double wing" / DNA binding motif / VIRAL PROTEIN-DNA complex VIRAL PROTEIN/DNA / MotA / dsDNA / "Double wing" / DNA binding motif / VIRAL PROTEIN-DNA complex | ||||||
| Function / homology |  Function and homology information Function and homology information | ||||||
| Biological species |  Enterobacteria phage T4 (virus) Enterobacteria phage T4 (virus)synthetic construct (others) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.955 Å | ||||||
 Authors Authors | Cuypers, M.G. / Robertson, R.M. / Knipling, L. / Hinton, D.M. / White, S.W. | ||||||
 Citation Citation | Journal: Nucleic Acids Res. / Year: 2018 Title: The phage T4 MotA transcription factor contains a novel DNA binding motif that specifically recognizes modified DNA. Authors: Cuypers, M.G. / Robertson, R.M. / Knipling, L. / Waddell, M.B. / Moon, K. / Hinton, D.M. / White, S.W. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 5jlt.cif.gz | 161.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb5jlt.ent.gz | 121.4 KB | Display | PDB format |
| PDBx/mmJSON format | 5jlt.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/jl/5jltftp://data.pdbj.org/pub/pdb/validation_reports/jl/5jlt | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1kafS S: Starting model for refinement |
|---|---|
| Similar structure data |








-Links
 PDBj
PDBj






































- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| 3 | 
| ||||||||
| 4 | 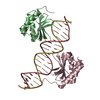
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 14548.928 Da / Num. of mol.: 4 Source method: isolated from a genetically manipulated source Details: non-native amino acids from expression vector= EGDIHM. N-TERM RESIDUES (93-96) = ELLK. LINKER (97-104) = KRATRKAR. HTTP://WWW.UNIPROT.ORG/UNIPROT/P22915 Source: (gene. exp.) Enterobacteria phage T4 (virus) / Gene: motA / Production host:  Escherichia coli (E. coli) / Strain (production host): BL21(DE3) / References: UniProt: P22915 Escherichia coli (E. coli) / Strain (production host): BL21(DE3) / References: UniProt: P22915#2: DNA chain | Mass: 6710.366 Da / Num. of mol.: 2 / Source method: obtained synthetically / Source: (synth.) synthetic construct (others) #3: DNA chain | Mass: 6790.414 Da / Num. of mol.: 2 / Source method: obtained synthetically / Source: (synth.) synthetic construct (others) #4: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 387 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 387 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.47 Å3/Da / Density % sol: 53.5 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, hanging drop / pH: 6.5 Details: 23% PEG 8K, 0.1 M Na Acetate, 0.1 M NaCacodylate, pH 6.5, and 3% glycerol |
-Data collection
| Diffraction | Mean temperature: 100 K / Ambient temp details: cryostream |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 22-ID / Wavelength: 1 Å / Beamline: 22-ID / Wavelength: 1 Å |
| Detector | Type: MAR CCD 130 mm / Detector: CCD / Date: Jun 25, 2014 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 2.955→93.12 Å / Num. obs: 17332 / % possible obs: 100 % / Redundancy: 23.4 % / Biso Wilson estimate: 65.2 Å2 / CC1/2: 0.999 / Rmerge(I) obs: 0.167 / Net I/σ(I): 15.1 |
| Reflection shell | Resolution: 2.955→3.11 Å / Redundancy: 23.5 % / Mean I/σ(I) obs: 2 / % possible all: 100 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 1KAF + DNA helix from COOT Resolution: 2.955→62.588 Å / Cross valid method: FREE R-VALUE / σ(F): 1.42 / Phase error: 35.46 Details: The settings of PHENIX.REFINE were tuned to use reference model restraints (PDB: 1KAF), NCS and the twin law K,H,-L.
| |||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å | |||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 65.2 Å2 | |||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.955→62.588 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
|
