 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-5hsq: The surface engineered photosensory module (PAS-GAF-PHY) of the b... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 5hsq | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | The surface engineered photosensory module (PAS-GAF-PHY) of the bacterial phytochrome Agp1 (AtBphP1) in the Pr form, chromophore modelled with an endocyclic double bond in pyrrole ring A. | ||||||||||||
 Components Components | Bacteriophytochrome protein | ||||||||||||
 Keywords Keywords |  SIGNALING PROTEIN / surface mutations / bilin chromophore / photoisomerization SIGNALING PROTEIN / surface mutations / bilin chromophore / photoisomerization | ||||||||||||
| Function / homology |  Function and homology information Function and homology informationred or far-red light photoreceptor activity / red, far-red light phototransduction / protein histidine kinase activity / detection of visible light / histidine kinase / phosphorelay sensor kinase activity / regulation of DNA-templated transcriptionSimilarity search - Function | ||||||||||||
| Biological species |  Agrobacterium fabrum (bacteria) Agrobacterium fabrum (bacteria) | ||||||||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.85 Å | ||||||||||||
 Authors Authors | Nagano, S. / Scheerer, P. / Zubow, K. / Lamparter, T. / Krauss, N. | ||||||||||||
| Funding support |  United Kingdom, United Kingdom,  Germany, 3items Germany, 3items
| ||||||||||||
 Citation Citation | Journal: J.Biol.Chem. / Year: 2016 Title: The Crystal Structures of the N-terminal Photosensory Core Module of Agrobacterium Phytochrome Agp1 as Parallel and Anti-parallel Dimers. Authors: Nagano, S. / Scheerer, P. / Zubow, K. / Michael, N. / Inomata, K. / Lamparter, T. / Krau, N. | ||||||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 5hsq.cif.gz | 216.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb5hsq.ent.gz | 170.9 KB | Display | PDB format |
| PDBx/mmJSON format | 5hsq.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/hs/5hsqftp://data.pdbj.org/pub/pdb/validation_reports/hs/5hsq | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  5i5lC  2veaS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |








-Links
 PDBj
PDBj







- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
| ||||||||
| Components on special symmetry positions |
|
-Components
-Protein , 1 types, 1 molecules A
| #1: Protein | Mass: 54736.945 Da / Num. of mol.: 1 / Mutation: E86A, E87A, E336A, K337A Source method: isolated from a genetically manipulated source Details: The residue numbering in the PDB entry differs from the one shown in this sequence alignment. The first residue in the PDB entry is 'Leu17' in accordance with the sequence for the entry ...Details: The residue numbering in the PDB entry differs from the one shown in this sequence alignment. The first residue in the PDB entry is 'Leu17' in accordance with the sequence for the entry AAT99575 (NCBI). The respective numbering was used in a number of publications on this protein, and we did not change it in this entry for the sake of consistency with published work and because the true N-terminus has not yet been identified at the protein level. Source: (gene. exp.) Agrobacterium fabrum (bacteria) / Strain: C58 / ATCC 33970 / Gene: Atu1990 / Plasmid: pET21b(+)Details (production host): The coding sequence of the protein of interest was subcloned using NdeI and XhoI sites of the plasmid. Production host: Escherichia coli BL21(DE3) (bacteria) / References: UniProt: Q7CY45 |
|---|
-Non-polymers , 5 types, 353 molecules 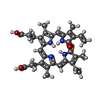




| #2: Chemical | ChemComp-BLA /  Mass: 582.646 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C33H34N4O6 Mass: 582.646 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C33H34N4O6 |
|---|---|
| #3: Chemical | ChemComp-CA /  Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca |
| #4: Chemical | ChemComp-GOL / Glycerol Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3 |
| #5: Chemical | ChemComp-IMD / Imidazole Mass: 69.085 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H5N2 Mass: 69.085 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H5N2 |
| #6: Water | ChemComp-HOH / WaterMass: 18.015 Da / Num. of mol.: 349 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.59 Å3/Da / Density % sol: 52.6 % / Description: Bipyramidal |
|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, hanging drop / pH: 6.5 Details: 0.03 M diethyleneglycol, 0.03 M triethyleneglycol, 0.03 M tetraethyleneglycol, 0.03 M pentaethyleneglycol, 20% glycerol, 10% PEG8000, 0.05 M MES, 0.05 M imidazole pH 6.5 |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: ID14-4 / Wavelength: 1.0718 Å / Beamline: ID14-4 / Wavelength: 1.0718 Å |
| Detector | Type: ADSC QUANTUM 315r / Detector: CCD / Date: Sep 8, 2011 |
| Radiation | Monochromator: Si(111) / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.0718 Å / Relative weight: 1 |
| Reflection | Resolution: 1.85→34.67 Å / Num. all: 49518 / Num. obs: 49518 / % possible obs: 98.6 % / Redundancy: 4.8 % / Biso Wilson estimate: 28.9 Å2 / Rmerge(I) obs: 0.048 / Net I/σ(I): 18.2 |
| Reflection shell | Resolution: 1.85→1.95 Å / Redundancy: 4.7 % / Rmerge(I) obs: 0.56 / Mean I/σ(I) obs: 2.8 / % possible all: 99.8 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 2)9C, 2VEA (PHY domain) Resolution: 1.85→34.31 Å / Cor.coef. Fo:Fc: 0.963 / Cor.coef. Fo:Fc free: 0.948 / SU B: 6.454 / SU ML: 0.098 / Cross valid method: THROUGHOUT / ESU R: 0.13 / ESU R Free: 0.127 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 37.842 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: 1 / Resolution: 1.85→34.31 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|