 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-5eje: Crystal structure of E. coli Adenylate kinase G56C/T163C double m... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 5eje | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Crystal structure of E. coli Adenylate kinase G56C/T163C double mutant in complex with Ap5a | |||||||||
 Components Components | Adenylate kinase | |||||||||
 Keywords Keywords | TRANSFERASE / Adenylate kinase / G56C and T163C variant / disulfide bond / Ap5A ligand | |||||||||
| Function / homology |  Function and homology information Function and homology informationpurine ribonucleotide interconversion / ADP biosynthetic process / nucleoside monophosphate metabolic process / nucleoside diphosphate metabolic process / adenylate kinase / adenylate kinase activity / AMP salvage / nucleoside diphosphate kinase activity / AMP binding / phosphorylation ...purine ribonucleotide interconversion / ADP biosynthetic process / nucleoside monophosphate metabolic process / nucleoside diphosphate metabolic process / adenylate kinase / adenylate kinase activity / AMP salvage / nucleoside diphosphate kinase activity / AMP binding / phosphorylation / magnesium ion binding / ATP binding / cytosol / cytoplasmSimilarity search - Function | |||||||||
| Biological species |  Escherichia coli (E. coli) Escherichia coli (E. coli) | |||||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.9 Å | |||||||||
 Authors Authors | Sauer, U.H. / Kovermann, M. / Grundstrom, C. / Wolf-Watz, M. / Sauer-Eriksson, A.E. | |||||||||
| Funding support |  Sweden, 2items Sweden, 2items
| |||||||||
 Citation Citation | Journal: Proc. Natl. Acad. Sci. U.S.A. / Year: 2017 Title: Structural basis for ligand binding to an enzyme by a conformational selection pathway. Authors: Kovermann, M. / Grundstrom, C. / Sauer-Eriksson, A.E. / Sauer, U.H. / Wolf-Watz, M. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 5eje.cif.gz | 186.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb5eje.ent.gz | 150.1 KB | Display | PDB format |
| PDBx/mmJSON format | 5eje.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ej/5ejeftp://data.pdbj.org/pub/pdb/validation_reports/ej/5eje | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  4akeS S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
| ||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | / AK / ATP-AMP transphosphorylase / ATP:AMP phosphotransferase / Adenylate monophosphate kinase Mass: 23668.160 Da / Num. of mol.: 2 / Mutation: G56C and T163C Source method: isolated from a genetically manipulated source Source: (gene. exp.) Escherichia coli (E. coli) / Strain: E24377A / ETEC / Gene: adk, EcE24377A_0513 / Production host: Escherichia coli (E. coli)References: UniProt: A7ZIN4, UniProt: P69441*PLUS, adenylate kinase#2: Chemical | 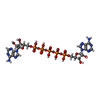  Mass: 916.367 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C20H29N10O22P5 Mass: 916.367 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C20H29N10O22P5#3: Chemical |   Mass: 58.933 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Co Mass: 58.933 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Co#4: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 552 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 552 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.49 Å3/Da / Density % sol: 50.69 % |
|---|---|
| Crystal grow | Temperature: 291 K / Method: vapor diffusion, hanging drop / pH: 5.8 Details: Purified AdK in 50 mM NaCl and 30 mM MES buffer, pH 6.0 was concentrated to 13 mg/ml and co-crystallized with a 5 molar excess of Ap5a. A typical drop contained 1 microL of protein mixed ...Details: Purified AdK in 50 mM NaCl and 30 mM MES buffer, pH 6.0 was concentrated to 13 mg/ml and co-crystallized with a 5 molar excess of Ap5a. A typical drop contained 1 microL of protein mixed with 1 microL of precipitant and equilibrated against 1 mL reservoir solution containing 26-28% PEG 8K, 10 mM CoCl2 and 0.1 M NaOAc, pH 5.8). |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: MAX II / Beamline: I911-2 / Wavelength: 1.038 Å |
| Detector | Type: MAR CCD 165 mm / Detector: CCD / Date: Sep 19, 2015 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.038 Å / Relative weight: 1 |
| Reflection | Resolution: 1.9→26.8 Å / Num. obs: 37833 / % possible obs: 99.6 % / Redundancy: 14.6 % / Rmerge(I) obs: 0.12 / Net I/σ(I): 18.8 |
| Reflection shell | Resolution: 1.9→1.97 Å / Redundancy: 14.1 % / Rmerge(I) obs: 1.99 / Mean I/σ(I) obs: 1.8 / % possible all: 97.5 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 4AKE Resolution: 1.9→26.8 Å / SU ML: 0.26 / Cross valid method: FREE R-VALUE / Phase error: 25.76 / Stereochemistry target values: ML
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.9→26.8 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
|
