 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-5dd7: Structure of thiamine-monophosphate kinase from Acinetobacter bau... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 5dd7 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Structure of thiamine-monophosphate kinase from Acinetobacter baumannii in complex with AMPPNP and thiamine-monophosphate | ||||||
 Components Components | Thiamine-monophosphate kinase | ||||||
 Keywords Keywords |  TRANSFERASE / SSGCID / Structural Genomics / Seattle Structural Genomics Center for Infectious Disease TRANSFERASE / SSGCID / Structural Genomics / Seattle Structural Genomics Center for Infectious Disease | ||||||
| Function / homology |  Function and homology informationthiamine-phosphate kinase / thiamine-phosphate kinase activity / thiamine diphosphate biosynthetic process / thiamine biosynthetic process / phosphorylation / magnesium ion binding / ATP binding Function and homology informationthiamine-phosphate kinase / thiamine-phosphate kinase activity / thiamine diphosphate biosynthetic process / thiamine biosynthetic process / phosphorylation / magnesium ion binding / ATP bindingSimilarity search - Function | ||||||
| Biological species |  Acinetobacter baumannii (bacteria) Acinetobacter baumannii (bacteria) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.7 Å | ||||||
 Authors Authors | Seattle Structural Genomics Center for Infectious Disease (SSGCID) | ||||||
 Citation Citation | Journal: Sci Rep / Year: 2019 Title: Crystal structures of thiamine monophosphate kinase from Acinetobacter baumannii in complex with substrates and products. Authors: Sullivan, A.H. / Dranow, D.M. / Horanyi, P.S. / Lorimer, D.D. / Edwards, T.E. / Abendroth, J. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 5dd7.cif.gz | 267.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb5dd7.ent.gz | 210.9 KB | Display | PDB format |
| PDBx/mmJSON format | 5dd7.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/dd/5dd7ftp://data.pdbj.org/pub/pdb/validation_reports/dd/5dd7 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  5cc8C  5cm7C  6mfmC  5d9u C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data | |
| Other databases |










-Links
 PDBj
PDBj











- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Components on special symmetry positions |
|
-Components
-Protein , 1 types, 2 molecules AB
| #1: Protein | Mass: 34959.656 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Acinetobacter baumannii (bacteria) / Gene: thiL, ABUW_0092 / Plasmid: AcbaC.17905.a.B1 / Production host: Escherichia coli (E. coli) / Strain (production host): BL21(DE3) / References: UniProt: A0A0D5YC82, thiamine-phosphate kinase |
|---|
-Non-polymers , 6 types, 513 molecules 



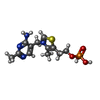

| #2: Chemical |  Mass: 506.196 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H17N6O12P3 / Comment: AMP-PNP, energy-carrying molecule analogue*YM Mass: 506.196 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H17N6O12P3 / Comment: AMP-PNP, energy-carrying molecule analogue*YM#3: Chemical | ChemComp-MG /  Mass: 24.305 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: Mg#4: Chemical | ChemComp-K /  Mass: 39.098 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: K Mass: 39.098 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: K#5: Chemical | Ethylene glycol Mass: 62.068 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C2H6O2 Mass: 62.068 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C2H6O2#6: Chemical |  Mass: 345.334 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C12H18N4O4PS Mass: 345.334 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C12H18N4O4PS#7: Water | ChemComp-HOH / | WaterMass: 18.015 Da / Num. of mol.: 493 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.17 Å3/Da / Density % sol: 43 % |
|---|---|
| Crystal grow | Temperature: 290 K / Method: vapor diffusion, sitting drop / pH: 7.5 Details: Microlytic MGCS G4 optimization screen: 19% PEG 3350, 200mM KNa-tartrate, 100mM HEPES/NaOH pH 7.75; AcbaC.17905.a.B1.PW37686 at 30mg/ml with 5mM MgCl2/ANP, then over night soak with in ...Details: Microlytic MGCS G4 optimization screen: 19% PEG 3350, 200mM KNa-tartrate, 100mM HEPES/NaOH pH 7.75; AcbaC.17905.a.B1.PW37686 at 30mg/ml with 5mM MgCl2/ANP, then over night soak with in reservoir solution with 5mM MgCl2/ANP/TMP; cryo: soak solution with 20% EG; tray 264486e7, puck RUX3-7 |
-Data collection
| Diffraction | Mean temperature: 100 K | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 21-ID-F / Wavelength: 0.97872 Å / Beamline: 21-ID-F / Wavelength: 0.97872 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: RAYONIX MX-225 / Detector: CCD / Date: Aug 7, 2015 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Monochromator: Diamond [111] / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 0.97872 Å / Relative weight: 1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 1.7→50 Å / Num. all: 66411 / Num. obs: 66088 / % possible obs: 99.5 % / Observed criterion σ(I): -3 / Redundancy: 6.2 % / Biso Wilson estimate: 18.91 Å2 / Rmerge F obs: 0.999 / Rmerge(I) obs: 0.053 / Rrim(I) all: 0.058 / Χ2: 0.95 / Net I/σ(I): 22.56 / Num. measured all: 407322 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell | Diffraction-ID: 1 / Rejects: 0
|
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 5d9u 5d9u Resolution: 1.7→39.41 Å / SU ML: 0.13 / Cross valid method: FREE R-VALUE / σ(F): 1.34 / Phase error: 16.02 / Stereochemistry target values: ML
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 77.83 Å2 / Biso mean: 26.7195 Å2 / Biso min: 10.37 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 1.7→39.41 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Total num. of bins used: 14
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
