 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4xyp | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of a piscine viral fusion protein | ||||||
 Components Components | Fusion protein | ||||||
 Keywords Keywords | VIRAL PROTEIN / heptad repeat / 6-helix bundle / class I viral fusion protein | ||||||
| Function / homology | Fusion glycoprotein F0, Isavirus / Fusion glycoprotein F0, Isavirus / membrane => GO:0016020 / 3,7-BIS(DIMETHYLAMINO)PHENOTHIAZIN-5-IUM / Fusion protein / Fusion protein Function and homology information Function and homology information | ||||||
| Biological species |  Infectious salmon anemia virus Infectious salmon anemia virus | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 2.1 Å | ||||||
 Authors Authors | Cook, J.D. / Lee, J.E. | ||||||
| Funding support |  Canada, 1items Canada, 1items
| ||||||
 Citation Citation | Journal: J.Biol.Chem. / Year: 2015 Title: Electrostatic Architecture of the Infectious Salmon Anemia Virus (ISAV) Core Fusion Protein Illustrates a Carboxyl-Carboxylate pH Sensor. Authors: Cook, J.D. / Soto-Montoya, H. / Korpela, M.K. / Lee, J.E. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4xyp.cif.gz | 38.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4xyp.ent.gz | 26.6 KB | Display | PDB format |
| PDBx/mmJSON format | 4xyp.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/xy/4xypftp://data.pdbj.org/pub/pdb/validation_reports/xy/4xyp | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|










-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| |||||||||
| Unit cell |
| |||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | Mass: 7912.075 Da / Num. of mol.: 1 / Fragment: UNP residues 156-225 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Infectious salmon anemia virus / Plasmid: pET46 / Production host:  Escherichia coli (E. coli) / Strain (production host): BL21(DE3) / References: UniProt: V5RD79, UniProt: C6F339*PLUS Escherichia coli (E. coli) / Strain (production host): BL21(DE3) / References: UniProt: V5RD79, UniProt: C6F339*PLUS | ||
|---|---|---|---|
| #2: Chemical | ChemComp-MBT / Methylene blue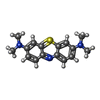  Mass: 284.399 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C16H18N3S / Comment: medication*YM Mass: 284.399 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C16H18N3S / Comment: medication*YM | ||
| #3: Chemical | Chloride  Mass: 35.453 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Cl Mass: 35.453 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Cl#4: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 6 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 6 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.6 Å3/Da / Density % sol: 52.62 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, sitting drop / pH: 4.5 Details: 1:1 volume ratio of 30mg/mL protein solution and 0.2 M lithium sulfate, 0.1 M sodium acetate, pH 4.5, 2 mg/mL tetramethylthionine chloride and 50% (v/v) PEG 400 |
-Data collection
| Diffraction | Mean temperature: 100 K | |||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: CLSI / Beamline: 08ID-1 / Wavelength: 0.97949 Å | |||||||||||||||||||||||||||
| Detector | Type: RAYONIX MX-300 / Detector: CCD / Date: Dec 14, 2013 / Details: 9 CCDs, 9 tiled fiber-optic tapers | |||||||||||||||||||||||||||
| Radiation | Monochromator: DOUBLE CRYSTAL MONOCHROMATOR / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | |||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 0.97949 Å / Relative weight: 1 | |||||||||||||||||||||||||||
| Reflection | Resolution: 2.1→38.71 Å / Num. obs: 5037 / % possible obs: 97.6 % / Redundancy: 4.7 % / Biso Wilson estimate: 33.75 Å2 / CC1/2: 0.999 / Rmerge(I) obs: 0.066 / Rpim(I) all: 0.032 / Net I/σ(I): 13.7 / Num. measured all: 23796 | |||||||||||||||||||||||||||
| Reflection shell | Diffraction-ID: 1 / Rejects: 0
|
-Phasing
| Phasing | Method: molecular replacement | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Phasing MR |
|
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 2.1→38.707 Å / SU ML: 0.19 / Cross valid method: THROUGHOUT / σ(F): 1.93 / Phase error: 23.23 / Stereochemistry target values: ML
| |||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | |||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 101.88 Å2 / Biso mean: 45.5257 Å2 / Biso min: 23.55 Å2 | |||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 2.1→38.707 Å
| |||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Total num. of bins used: 4
|
